新型苯甘氨酸衍生物“刷型”手性固定相的制备及其应用*
2015-04-23付克勤张俊俊柏正武
宾 琴,付克勤,张俊俊,陈 伟,柏正武
(武汉工程大学化学与环境工程学院,湖北武汉 430073)
手性药物中各对映体间的药理活性通常具有较大差异[1],往往只有一种对映体有药理活性,而另一种对映体无药理活性,甚至有毒副作用[2-4]。随着人们对手性药物需求的不断增长,研究手性化合物对映体的拆分与检测具有重要意义。
手性固定相的高效液相色谱法是检测手性化合物对映体最成熟的方法[5]。“刷型”固定相的选择体为手性小分子,这类固定相具有柱容量高、对映体选择性好等优点,常用于氨基酸及氨基酸衍生物或内酰胺等手性化合物的对映体检测中。苯甘氨酸衍生物常用作“刷型”固定相的选择体[6-11]。Pirkle 等[9]研究了一类苯甘氨酸衍生物手性固定相,虽然其手性识别能力不及Whelk-O1,但可以识别的化合物种类多,如非甾体抗炎药、苯二氮杂酮等,应用也较为广泛;袁黎明等[10-11]报道了两种 D-苯甘氨酸衍生物手性固定相,这类固定相的分离选择性倾向于醇类、胺类、氨基酸的对映异构体以及一些手性药物。因此,研究这类固定相的制备方法,特别是手性选择体的固定化方法,发掘新的“刷型”手性固定相具有重要的实用价值。
本文以氨基酸(1a~1c)为原料,先制得氨基酸甲酯盐酸盐(2a~2c);2再与3,5-二硝基苯甲酰氯(3)反应合成了3个N-酰基-D-苯甘氨酸甲酯(4a ~4c,Scheme 1)。D-2-氯苯甘氨酸(1b)和3,5-二甲基苯甲酰氯(5)或 D-4-氯苯甘氨酸(1d)和3分别经缩合反应合成了N-3,5-二甲基苯甲酰基-D-2-氯苯甘氨酸(6)或 N-3,5-二硝基苯甲酰基-D-4-氯苯甘氨酸(7,Scheme 2);其中 4b,4c,6和7为新化合物,其结构经1H NMR,IR和元素分析表征。分别以胺-酯交换法和缩合反应将4a,4c,6和7固定在氨丙基硅胶(P-NH2,Chart 1)表面,制得4种新型的“刷型”手性固 定 相 4a/P-NH2(简 称4a'),4c/P-NH2(4c'),6/P-NH2(6')和 7/P-NH2(7')。对两种固定化方法进行了比较研究。结果表明:以缩合法制备的6'和7'的手性选择体负载量相对较高,分别为 0.24 mmol·g-1和 0.33 mmol·g-1。以胺-酯交换法制备的 4a'和 4c'的负载量分别为 0.11 mmol·g-1和 0.10 mmol·g-1。用4-二甲基氨基吡啶(DMAP)和1-羟基苯并三唑(HOBt)为催化剂能避免4a~4c在固定化反应中的消旋化。初步评价了6'对8种手性化合物(A~H,Chart 2)的手性识别性能。
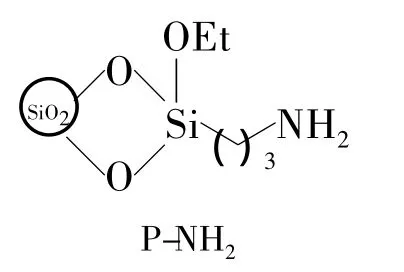
Chart 1
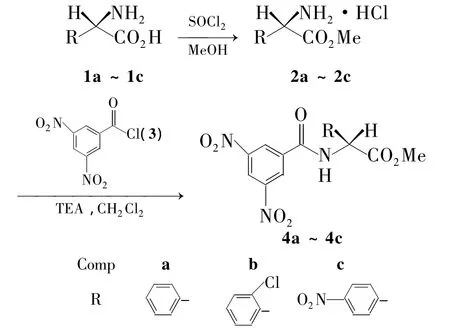
Scheme 1
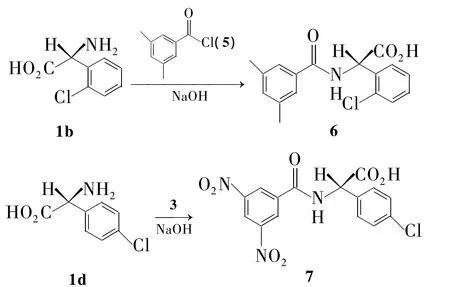
Scheme 2
1 实验部分
1.1 仪器与试剂
Varian Mercury VX-300M 型核磁共振仪(CDCl3为溶剂,TMS 为内标);Nicolet 5DX FT-IR型红外光谱仪(KBr压片);Vario EL III CHNOS型元素分析仪;Alltech 1666型色谱柱填充泵;250 mm×4.6 mm不锈钢空色谱柱;Waters型高效液相色谱仪系统。
2a ~2c 参照文献[12-13]方法合成,其表征数据[14]与 Scheme 1预期结构吻合;球形硅胶(5 μm),青岛美高化工有限公司;1a~1d,北京红杉精化科技有限公司;HOBt和 2-乙氧基-1-乙氧碳酰基-1,2-二氢喹啉(EEDQ),苏州昊帆生物科技有限公司;其余所用试剂均为分析纯。
1.2 合成
(1)4a~4c的合成(以4a为例)
三口烧瓶中依次加入D-苯甘氨酸甲酯盐酸盐(2a)9.28 g(46 mmol),二氯甲烷400 mL和三乙胺12.5 mL(90 mmol),搅拌使其溶解;加入3 9.68 g(42 mmol),于20 ℃反应24 h。过滤,滤液依次用5%NaCO3溶液(3×100 mL)和饱和NaCl溶液(3×100 mL)洗涤,无水硫酸镁干燥,蒸除溶剂后用二氯甲烷-正己烷重结晶两次得4a。
用类似的方法合成4b和4c。
N-3,5-二硝基苯甲酰基-D-苯甘氨酸甲酯(4a):白色絮状固体,产率81%,m.p.179℃ ~7.43 ~7.44(m,5H,ArH),5.79(d,J=7.5 Hz,1H,CH),3.82(s,3H,CH3);IR ν:3 345(CONH),1 736(CO2),1 648(CONH),1 217(C -O -C)cm-1;Anal.calcd for C16H13N3O7:C 53.49,H 3.65,N 11.70;found C 53.68,H 3.67,N 11.73。
N-3,5-二硝基苯甲酰基-D-2-氯苯甘氨酸甲酯(4b):淡黄色固体,产率73%,m.p.133℃ ~135℃,+86.2°(c 0.4,THF);1H NMR(DMSO-d6)δ:10.05(d,J=6.3 Hz,1H,CONH),7.45 ~9.13(m,7H,ArH),6.10(d,J=6.0 Hz,1H,CH),3.73(s,3H,CH3);IR ν:3 283(CONH),1 717(CO2),1 650(CONH),1 234(C -O -C)cm-1;Anal.calcd for C16H12N3O7Cl:C 48.81,H 3.07,N 10.67;found C 48.86,H 3.16,N 10.54。
N-3,5-二硝基苯甲酰基-D-4-硝基苯甘氨酸甲酯(4c):淡棕色固体,产率88%,m.p.166℃ ~168 ℃,0°(c 0.4,THF);1H NMR δ:7.59 ~9.20(m,7H,ArH),8.33(s,1H,CONH),5.95(d,J=6.9 Hz,1H,CH),3.86(s,3H,CH3);IR ν:3 341(CONH),1 744(CO2),1 644(CONH),1 209(C - O - C)cm-1;Anal.calcd for C16H12N4O9:C 47.53,H 2.99,N 13.86;found C 47.74,H 3.14,N 13.76。
(2)N-酰基-D-苯甘氨酸(6 和 7)的合成
在三口烧瓶中加入3,5-二甲基苯甲酸53 g和二氯亚砜150 mL,搅拌下回流反应3 h。减压蒸馏,收集105℃/2.7 kPa馏分得无色透明液体5 54 g,产率 92%。
在三口烧瓶中加入1b 9.3 g(50 mmol)和2 mol·L-1NaOH溶液190 mL,搅拌使其溶解。冷却至-2℃,缓慢滴加5 14.8 mL(100 mmol),滴毕,于-2℃反应24 h。过滤,滤液用乙醚(3×50 mL)萃取,收集水相,缓慢滴加6 mol·L-1盐酸至pH 2。用乙酸乙酯(3×50 mL)萃取,合并乙酸乙酯层,用无水硫酸镁干燥,蒸除溶剂后用乙酸乙酯-正己烷重结晶两次得6。
用1d[8.0 g(43 mmol)]替代1b ,用 3[21.0 g(91 mmol)]替代5,用类似的方法合成粗品,经活性碳脱色,用乙酸乙酯-正己烷重结晶得7。
6:白色晶体,产率54%,m.p.152℃ ~154℃,+59.0°(c 0.4,乙酸乙酯);1H NMR δ:7.13 ~ 7.49(m,7H,ArH),6.12(d,J=7.2 Hz,1H,CH),2.32(s,6H,CH3);IR ν:3 427(CONH,CO2H),3 311~2 636(CO2H),1 720(CO2H),1 628(CONH)cm-1;Anal.calcd for C17H16NO3Cl:C 64.26,H 5.18,N 4.41;found C 64.55,H 5.06,N 4.09。
7:白色晶体,产率31%,m.p.219℃ ~221℃,-67.1°(c 0.4,乙酸乙酯);1H NMR(DMSO-d6)δ:13.26(s,1H,CO2H),9.90(d,J=6.6 Hz,1H,CONH),7.47 ~ 9.13(m,7H,ArH),5.68(d,J=6.9 Hz,1H,CH);IR ν:3 307(NH,CO2H),3 202~2 680(CO2H),1 711(CO2H),1 658(CONH)cm-1;Anal.calcd for C15H10N3O7Cl·H2O:C 45.30,H 3.04,N 10.57;found C 45.42,H 2.82,N 10.75。
1.3 苯甘氨酸衍生物的固定化
(1)4a'和 4b'的合成
在两口瓶中依次加入4a 3.00 g(8.4 mmol),HOBt 1.35 g(10.0 mmol)和干燥 THF 27 mL,搅拌使其溶解;加入干燥氨丙基硅胶3.22 g和催化量的DMAP,于室温反应48 h。离心分离,沉淀用THF洗涤,真空干燥得白色固体4a'3.35 g,增重0.13 g,负载量 0.11 mmol·g-1。滤液滴至正己烷中,析出沉淀,过滤,滤饼干燥,测得+93.7°(c 0.4,THF)。
用类似的方法合成4b',增重0.13 g,负载量为0.10 mmol·g-1。同法测得THF)。
(2)6'和7'的合成
在两口瓶中依次加入6 2.00 g(6.3 mmol),EEDQ 1.71 g(6.9 mmol)和干燥 THF 7 mL,搅拌使其溶解;加入干燥氨丙基硅胶3.41 g,于室温反应48 h。离心分离,沉淀用THF洗涤,真空干燥得白色固体 6'3.77 g,增重 0.36 g,负载量 0.33 mmol·g-1。滤液滴至正己烷中,析出沉淀,过滤,滤饼干燥,测得+57.3°(c 0.4,乙酸乙酯)。
用类似的方法制得7',增重0.32 g,负载量0.24 mmol·g-1。同法测得0°(c 0.4,乙酸乙酯)。
1.4 6'的对映体选择性检测
采用匀浆法填柱。用正己烷(30 mL)/异丙醇(95/5)匀浆,正己烷作顶替液,用填充泵在6 000 psi压力下将6'(3.1 g)填入不锈钢色谱柱中。以联苯为检测物,正己烷/异丙醇(90/10,V/V)为流动相测定柱效,用1,3,5-三叔丁基苯为样品测得死时间。以乙醇为溶剂,将手性化合物A ~H配成1 mg·mL-1溶液,并经0.2 μm 滤膜过滤。分别以异丙醇和乙醇与正己烷组成的混合溶液为正相流动相,以甲醇和乙腈与水组成的混合溶液为反相流动相,测试6'的对映体选择性。在所有检测中,流速均为1 mL·min-1,柱温为25℃。
2 结果与讨论
2.1 合成中的消旋化
在N-酰化反应过程中,为了尽量避免产物的消旋化,反应均在较低温度下进行。实验中发现,在合成4c时发生了消旋化,测得其比旋度为零。这是因为在4c的分子结构中,硝基是强吸电子基团,其对位的苄基碳原子在三乙胺的催化下可逆地生成碳负离子,这个碳负离子能够被酯基和苯基上的硝基所稳定。由于碳负离子属于sp3杂化,呈角锥形的结构,又因为碳负离子上的一对未成对电子体积小,此构型能自由翻转,从而发生消旋化;或者所生成的碳负离子转变为烯醇式的结构,也能发生消旋化。文献[15-19]对氨基酸在碱性介质中消旋化的现象进行了探讨,几乎都认为消旋化经历了碳负离子的过程,本文也认同这一观点。
2.2 苯甘氨酸衍生物的固定化反应
HOBt常用作酰胺键形成促进剂,能有效抑制产物消旋[20-21]。在 N-酰基-D-苯甘氨酸甲酯的固定化反应中(即4'的合成中),不加HOBt时,反应完成后从反应液中回收的原料有较大程度的消旋。而使用HOBt后,从反应混合物中回收的原料的比旋度略低于反应前原料的比旋度。一个可能的原因是为避免因加热带来的消旋化,将反应混合物滴入正己烷中,使原料沉淀出来,会有少许HOBt混杂在其中,所以比旋度稍低;另一个可能的原因就是确实发生了少量的消旋化。总的来说HOBt起到了阻止消旋化的作用。但在不同的反应中,其阻止消旋化的机理不尽相同。
推测本反应的机理是:4与DMAP形成的活性中间体(I)较易烯醇化,生成一对几何异构体II和III;当II和III可逆地返回到I时,部分构型发生了变化,从而产生消旋现象。在反应体系中加入HOBt后,I与其反应生成活性酯IV,与I相比IV不易烯醇化,IV与氨丙基硅胶(P-NH2)反应生成V(即为固定相4')(Scheme 3)。其实,PNH2上的氨基与HOBt都能和I反应,但HOBt溶于溶液中,与I之间的反应是均相的,而P-NH2与I之间的反应是异相反应,所以HOBt和I的反应快一些。因此,用胺-酯交换反应固定苯甘氨酸甲酯衍生物时以DMAP和HOBt共同作为催化剂较为合适。
根据P-NH2在固定化反应后质量的增量计算出固定相上手性选择体的负载量。用胺-酯交换反应固定4a和4b制得4a'和4b',其手性选择体的负载量分别为 0.11 mmol·g-1和 0.10 mmol·g-1。以EEDQ为缩合剂固定6时,6'负载量达0.33 mmol·g-1,但负载量并非越高越好。
用缩合法固定选择体时也可能发生消旋化。6和7分别从1b和1d制备,邻位的氯和对位的氯对苯基的电子效应没有很大区别。但7中的苯甲酰基有两个强吸电子的硝基。固定化反应是在室温下进行的,7在P-NH2的催化下,经过长时间的搅拌发生了消旋化。消旋化也应经过了形成碳负离子的过程,所形成的碳负离子得到硝基的稳定。相反,6中的苯甲酰基中含两个甲基,甲基不能稳定碳负离子,所以6在固定化时没有消旋化。
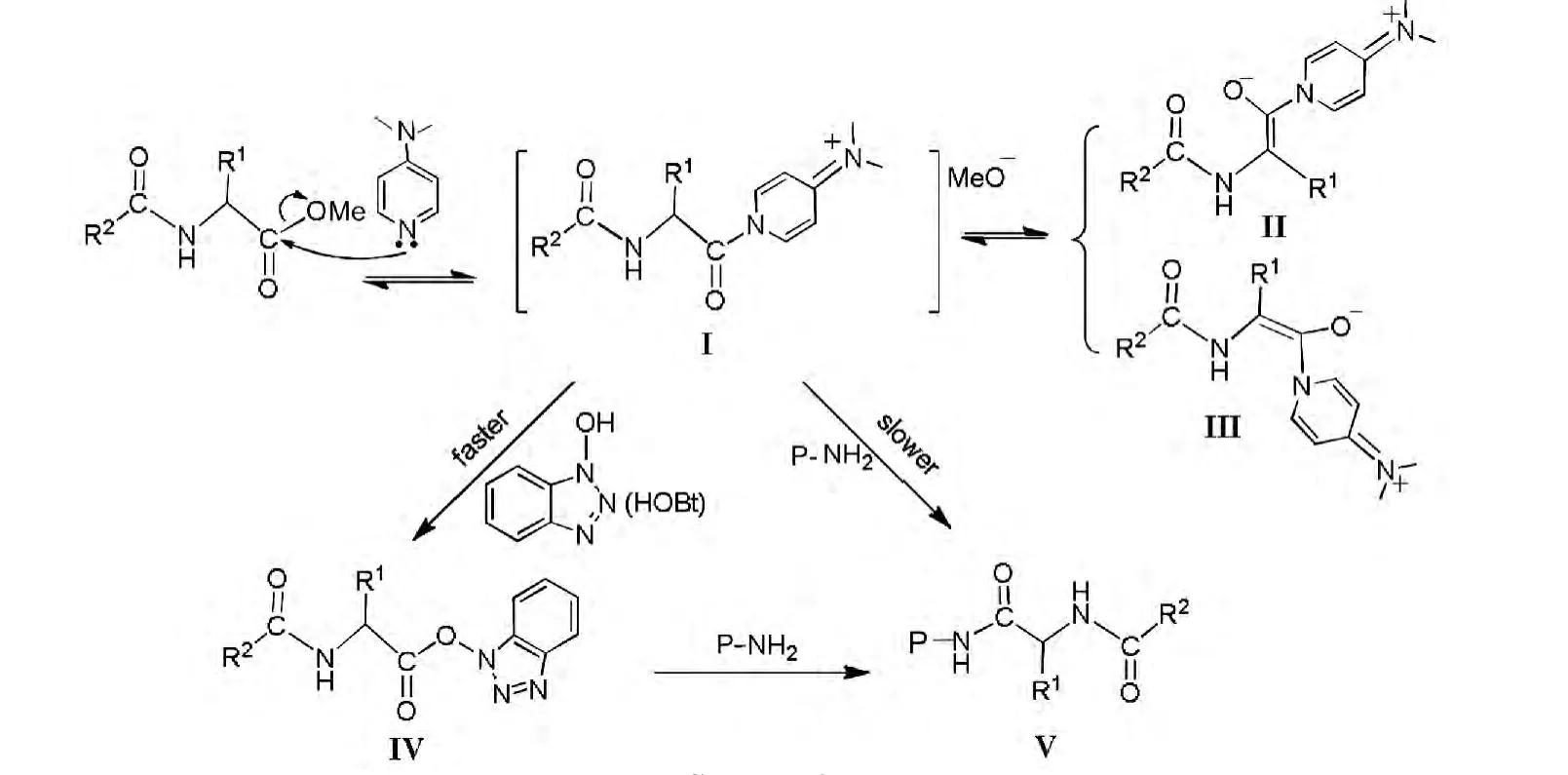
Scheme 3
2.3 6'的分离性能
在正己烷/异丙醇(90/10,V/V)为流动相时测得的柱效是27 100理论塔板数/米,对称因子为1.10。测得的死时间为3.196 min。用8种手性化合物(A~H)对6'的手性识别性能进行了初步检测,结果见表1。从表1可见,6'有较大的分离因子(α),即有较好的对映体选择性。但要提高分离度,还需要进一步摸索分离条件。
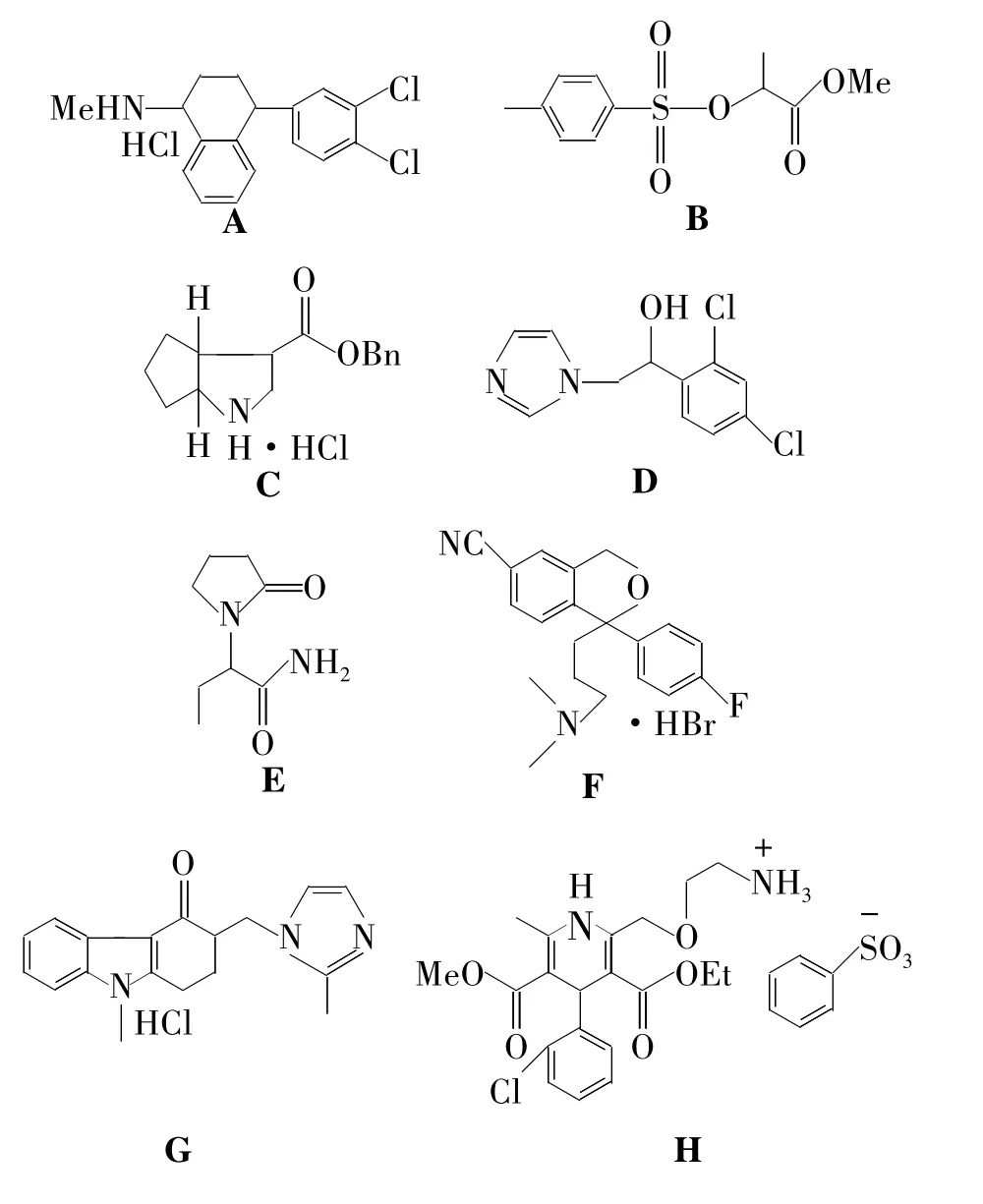
Chart 2
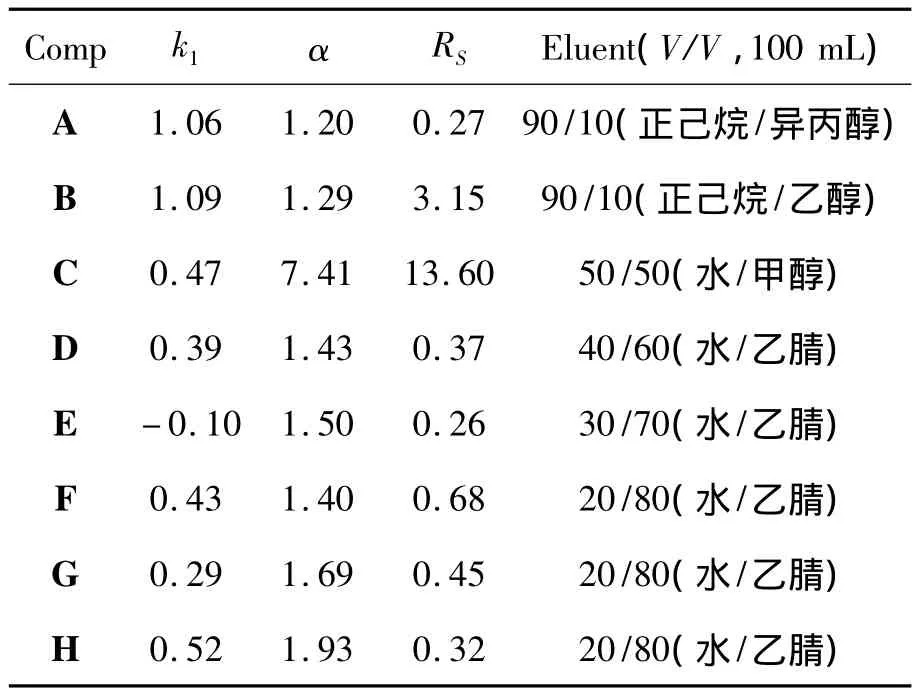
表1 A~H在6'上的色谱分离数据Table 1 Chromatographic data of A~H resolved by 6'
从表1还可知,6'在反相中有更好的手性识别性能,在乙腈/水的流动相中尤其如此。这可能是由于甲醇为质子溶剂,乙腈为非质子溶剂,而非质子溶剂与手性化合物的氢键作用比质子溶剂与手性化合物的氢键作用小,使得手性化合物与手性选择体之间有更好的相互作用,从而增强手性识别性能。
3 结论
合成了5个苯甘氨酸衍生物,并通过缩合反应及胺-酯交换反应将4种衍生物键合于氨丙基硅胶上,制备出4种新的“刷型”手性固定相。以胺-酯交换反应固定N-酰基-D-苯甘氨酸甲酯时,采用DMAP和HOBt为催化剂能有效避免反应过程中苯甘氨酸衍生物的消旋化;以EEDQ为缩合剂,固定 N-酰基-D-苯甘氨酸时,固定量相对较高,当苯甘氨酸的苯甲酰基上有吸电子基团时,在固定化过程中容易发生消旋化。
选择其中一种固定化方法能制得所需要负载量的固定相。对以 N-3,5-二甲基苯甲酰基-D-2-氯苯甘氨酸为手性选择体的固定相进行了手性识别性能评价,结果显示这种固定相在以乙腈/水为流动相的反相模式下有较好的对映体选择性。
[1]Gübitz G,Schmid M G.Chiral separation principles in chromatographic and electromigration techniques[J].Mol Biotechnol,2006,32:159 -179.
[2]Knoche B,Blaschke G.Investigations on the in vitro racemization of thalidomide by high-performance liquid chromatography[J].J Chromatogr A,1994,666:235 -240.
[3]Caldwell J.Importance of stereospecific bioanalytical monitoring in drug development[J].J Chromatogr A,1996,719:3 -13.
[4]Aboul-Enein H Y.High-performance liquid chromatographic enantioseparation of drugs containing multiple chiral centers on polysaccharide-type chiral stationary phases[J].J Chromatogr A,2001,906:185 -193.
[5]Chang Y X,Ren C X,Ruan Q,et al.Effect of single-walled carbon nanotubes on cellulose phenylcarbamate chiral stationary phases[J].Chem Res Chin Univ,2007,23:646 -649.
[6]Ward T J,Hamburg D M.Chiral separations[J].Anal Chem,2004,76:4635 -4644.
[7]Lämmerhofer M J.Chiral recognition by enantioselective liquid chromatography:Mechanisms and modern chiral stationary phases[J].J Chromatogr A,2010,(1217):814-856.
[8]Pirkle W H,House D W,Finn J M.Broad spectrum resolution of optical isomers using chiral high-performance liquid chromatographic bonded phases[J].J Chromatogr,1980,192:143 -158.
[9]Fernandes C,Palmeira A,Santos A,et al.Enantioresolution of chiral derivatives of xanthones on(S,S)-whelk-O1 and L-phenylglycine stationary phases and chiral recognition mechanism by docking approach for(S,S)-whelk-O1[J].Chirality,2013,25:89 -100.
[10]常银霞,周玲玲,向兰,等.D-苯甘氨酸手性固定相的制备及对多种手性化合物的拆分[J].分析测试学报,2007,26:107 -109.
[11]周玲玲,李国祥,王剑瑜,等.N-(4-甲基苯甲酰基)苯甘氨酸手性固定相的合成及对多种手性化合物的拆分[J].分析化学,2007,35:1301 -1304.
[12] 熊维,申东升,王国华.(+)-邻氯苯甘氨酸甲酯盐酸盐的合成工艺研究[J].化学试剂,2008,30:927-928.
[13]Kudelko A,Zieliński W,Ejsmont K.The reaction of optically activea-aminocarboxylic acid hydrazides with triethyl orthoesters[J].Tetrahedron,2011,67:7838 -7845.
[14]D-苯甘氨酸甲酯盐酸盐(2a):白色固体,产率90%,m.p.183 ℃ ~185 ℃,[α]25D+135.3°(c 0.4,CH3OH,下同);1H NMR(D2O)δ:7.47 ~7.54(m,5H,ArH),5.29(s,1H,CH),3.82(s,3H,CH3);IR ν:1 738(C=O),1 242(C - O - C)cm-1.D-2-氯苯甘氨酸甲酯盐酸盐(2b):无色晶体,产率87%,m.p.159 ℃ ~ 161 ℃,[α]25D+104.1°;1H NMR(D2O)δ:7.46 ~7.63(m,4H,ArH),5.70(s,1H,CH),3.85(s,3H,CH3);IR ν:1 756(C=O),1 188(C -O -C)cm-1.D-4-硝基苯甘氨酸甲酯盐酸盐(2c):淡黄色固体,产率70%,m.p.172 ℃ ~174 ℃,[α]25D-106.6°;1H NMR(D2O)δ:7.78 ~ 8.41(m,4H,ArH),5.56(s,1H,CH),3.86(s,3H,CH3);IR ν:1 744(C=O),1 250(C-O-C)cm-1.
[15]王家荣,何佺,彭阳峰.L-酪氨酸的消旋研究[J].高校化学工程学报,2007,21:141-145.
[16]方百盈,冯大炎.L-氨基酸消旋化作用的探讨[J].化学研究与应用,1997,9:40 -44.
[17]Ebbers E J,Ariaans G J A,Houbiers J P M,et al.Controlled racemization of optically active organic compounds:Prospects for asymmetric transformation[J].Tetrahedron,1997,53:9417 -9476.
[18]Smith G G,Sivakua T.Mechanism of the racemization of amino acids.Kinetics of racemization of arylglycines[J].J Org Chem,1983,48:627 -634.
[19]Neuberger A.Stereochemistry of amino acids[J].Adv Protein Chem,1948,4:297 -376.
[20]El-Faham A,Albericio F.Peptide coupling reagents,More than a letter soup[J].Chem Rev,2011,111:6557-6602.
[21]Carpino L A.1-Hydroxy-7-azabenzotriazole.An efficient peptide coupling additive[J].J Am Chem Soc,1993,115:4397 -4398.
