阿托伐他汀对ApoE基因敲除小鼠动脉粥样硬化病变中TRPC5表达的影响*
2015-04-15崔建国张清潭
齐 洁, 徐 芳, 马 慧, 崔建国, 张清潭△
(1滨州医学院附属医院老年内科,山东 滨州 256603; 2滨州医学院病理生理教研室,山东 烟台 264003)
阿托伐他汀对ApoE基因敲除小鼠动脉粥样硬化病变中TRPC5表达的影响*
齐 洁1, 徐 芳2, 马 慧1, 崔建国1, 张清潭1△
(1滨州医学院附属医院老年内科,山东 滨州 256603;2滨州医学院病理生理教研室,山东 烟台 264003)
目的: 观察载脂蛋白E基因敲除(ApoE-/-)小鼠动脉粥样硬化斑块形成过程中血管平滑肌细胞瞬时受体电位通道5(TRPC5)蛋白的表达变化,以及阿托伐他汀药物干预对TRPC5的影响并探讨其作用机制。方法: 将40只6周龄雄性ApoE-/-小鼠随机分为模型组和他汀干预组,高脂饲料喂养建立动脉粥样硬化模型。他汀干预组给予阿托伐他汀(20 mg·kg-1·d-1)灌胃,模型组给予等量生理盐水灌胃。20只同龄雄性野生型C57BL/6J小鼠给予普通饲料喂养作为正常对照组。各组小鼠分别喂养至20和30周龄,分别取10只取血并处死。取主动脉根部做石蜡切片行HE染色形态学观察,测量并计算斑块相对面积;免疫组织化学染色检测各组小鼠TRPC5蛋白表达变化。取胸腹段主动脉行实时荧光定量PCR,检测主动脉中TRPC5通道蛋白mRNA的表达水平。结果: 与模型组相比,阿托伐他汀干预组的血脂明显下降,斑块总面积明显减小,TRPC5蛋白水平及 mRNA含量明显下降;30周龄模型组的TRPC5蛋白表达稍高于20周龄模型组,但差异无统计学意义;30周干预组较20周干预组相比,TRPC5水平有降低趋势且差异有统计学意义。结论: 阿托伐他汀可能通过下调TRPC5蛋白表达从而延缓动脉粥样硬化进程。
TRPC5; 载脂蛋白E; 动脉粥样硬化; 血管平滑肌细胞; 阿托伐他汀
动脉粥样硬化(atherosclerosis,AS)是心脑血管疾病最常见的基本病因,严重影响人类的生活质量和健康水平[1]。其病理生理过程是由多种危险因子导致血管内皮损伤,刺激多种细胞(包括淋巴细胞、巨噬细胞、内皮细胞和血管平滑肌细胞等)参与局部脂质和糖类积聚、纤维组织增生和钙质沉着形成斑块,并伴有动脉血管中层退变。血管平滑肌细胞(vascular smooth muscle cells,VSMCs)增殖与凋亡是决定动脉粥样硬化斑块发生发展的重要环节[2-3]。其中,瞬时受体电位通道(transient receptor potential channel,TRPC)介导血管平滑肌细胞、血管内皮细胞、巨噬细胞等多种细胞参与了AS的病理生理过程[4],且已有研究发现TRPC家族成员之一的TRPC5蛋白表达主要位于血管平滑肌细胞,参与其收缩、肥大、增殖和迁移等生理及病理过程[5],能够促进AS的发展。本实验拟通过载脂蛋白E基因敲除(apolipoprotein E-knockout,ApoE-/-)小鼠建立AS模型,观察AS病程中不同时点主动脉VSMCs内TRPC5蛋白表达的改变,以及阿托伐他汀干预治疗后对该蛋白表达的影响,探讨阿托伐他汀抗AS的作用机制。
材 料 和 方 法
1 动物与试剂
实验组选取6周龄ApoE-/-雄性小鼠(品系C57BL/6J)40只,正常对照组为同周龄C57BL/6J雄性小鼠20只,均购自北京大学医学部实验动物中心,合格证编号为SCXK(京)2011-0012。小鼠体重18~20 g,饲养条件为二级,室温控制在23~25 ℃,相对湿度50%~60%,光照时间07:00~19:00,自由饮水及进食。阿托伐他汀由美国辉瑞公司惠赠(药物批号:国药准字J20070060),用0.9%氯化钠溶解。TRPC5抗体购自Abcam;SP试剂盒购自福州迈新生物技术开发有限公司;Trizol、反转录试剂盒和SYBR®Green荧光试剂盒为TaKaRa产品;其它相关试剂均由滨州医学院医药研究中心提供。
2 方法
2.1 动物分组与模型制备 选取40只6周龄ApoE-/-雄性小鼠给予高脂饮食(基础饲料78.85%+脂肪21%+胆固醇0.15%),随机分为模型组(生理盐水,0.1~0.2 mL,ig)和阿托伐他汀干预组(阿托伐他汀 20 mg·kg-1·d-1,ig),每组20只。同时选取20只同龄C57BL/6J雄性小鼠作为正常对照组,给予普通饲料饮食。
2.2 标本处理 每组小鼠喂养至20周龄和30周龄时各取10只处死,处死前禁食不禁水12 h。无菌条件下取出心脏和主动脉根部,4%多聚甲醛固定处理后石蜡包埋,制成4 μm连续切片用于HE染色以及TRPC5的免疫组织化学染色。分离出主动脉胸腹段于-80 ℃冻存,抽提主动脉总RNA,反转录后采用SYBR®Green实时荧光定量PCR检测TRPC5 mRNA的表达。
2.3 血清学指标测定 全血以2 500 r/min 4 ℃低温离心20 min,取血清,应用全自动生化分析仪检测甘油三酯(triglyceride,TG)、总胆固醇(total cholesterol,TC)、高密度脂蛋白胆固醇(high-density lipoprotein cholesterol,HDL-C)和低密度脂蛋白胆固醇(low-density lipoprotein cholesterol,LDL-C)含量。
2.4 病理组织学图像测量和分析 主动脉根部石蜡切片行HE染色,用Image-Pro Plus 6.0形态计量学测算斑块面积及血管内外膜长度,回归标准圆形后计算斑块面积与血管管腔面积相对比值。免疫组化染色检测TRPC5阳性染色积分吸光度值(integral absorbance,IA)(DAB显色为棕褐色)。PBS缓冲液代替Ⅰ抗作为空白对照。结果判定:光镜下,阳性表达的细胞围绕蓝色细胞核的胞浆呈棕褐色。每个标本共取5张切片染色,同一切片在相同环境下测量3次,取平均值作为测量结果。
2.5 RNA的提取和逆转录反应 采用Trizol两步法抽提主动脉的总RNA,按照TaKaRa的反转录试剂盒要求反转录提取总RNA,10 μL反应体系如下:RNA 2 μL,5×PrimeScript buffer 2 μL,PrimeScript RT Enzyme Mix I 0.5 μL,Oligo dT Prime(50 μmol/L) 0.5 μL,Random 6 mers(100 μmol/L) 0.5 μL,去RNA酶水 4.5 μL。反应条件为37 ℃ 15 min,85 ℃ 5 s,1个循环后最后得到的cDNA,-20 ℃保存备用。
2.6 Real-time PCR检测主动脉血管中TRPC5 mRNA的表达 采用实时荧光定量PCR检测TRPC5 mRNA的表达,内参照基因为GAPDH,由大连宝生物设计引物序列。TRPC5的上游引物为5’-ACAAAAAGGTCAACTACTCACCG-3’,下游引物为5’-CAGTGGCATAGTCCCCCTTCT-3’;GAPDH的上游引物为5’-AACTGCTTAGCACCCCTGGC-3’,下游引物为5’-ATGACCTTGCCCACACAGCCTT-3’。PCR反应体系如下:SYBR®Prime Ex Taq(Tli RNaseH Plus)10 μL,PCR上游引物(10 μmol/L) 0.4 μL,PCR下游引物(10 μmol/L) 0.4 μL,ROX Reference Dye(50×)0.4 μL,DNA模板 2 μL,dH2O 6.8 μL。反应条件为95 ℃ 30 s;然后95 ℃ 5 s,60 ℃ 34 s 共40个循环。反应结束,设定最佳阈值,得到Ct值。用2-ΔΔCt法计算。实验重复3次。
3 统计学处理
计量资料以均数±标准差(mean±SD)表示,所有统计分析均使用SPSS 13.0统计软件处理。组间均数比较采用单因素方差分析,两两比较采用q检验,以P<0.05为差异有统计学意义。
结 果
1 不同周龄各组小鼠血脂水平
ApoE-/-小鼠血脂可自发性升高,高脂饮食能加速血脂增高速度。与对照组比较,模型组血清的TC、TG和LDL-C均升高(P<0.05),以TC和LDL-C增高最为明显,且随周龄增加而增高;而HDL-C有所下降(P<0.05)。给予阿托伐他汀后血清TC、TG和LDL-C均降低(P<0.05),且以TC和LDL-C下降最为显著,HDL-C水平有所升高(P<0.05)。30周龄干预组与20周龄干预组比较,TC和LDL-C有下降趋势,HDL-C有升高趋势,但差异均无统计学意义,见表1。
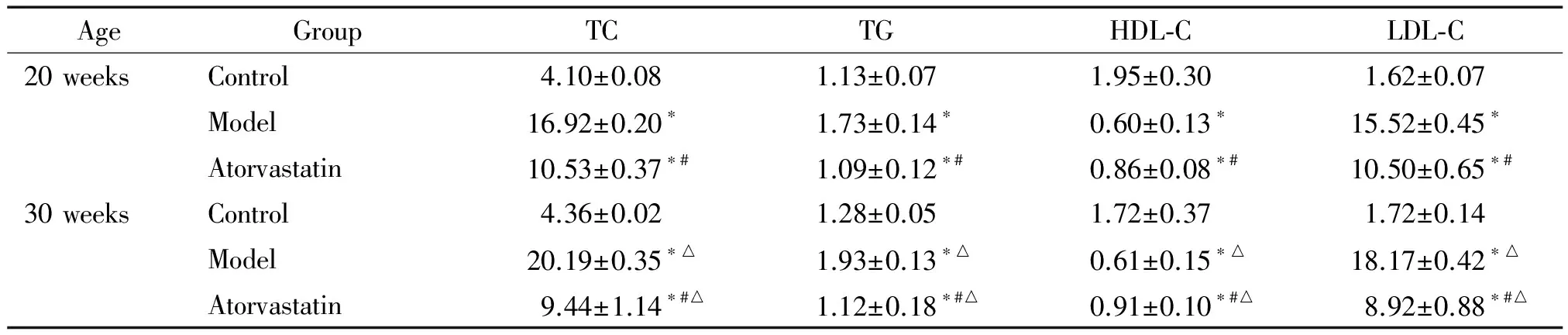
表1 各组小鼠血脂水平的比较
*P<0.05vscontrol at the same age;#P<0.05vsmodel at the same age;△P<0.05vs20 weeks in the same group.
2 不同周龄小鼠主动脉粥样硬化斑块的病理学观察
C57BL/6J小鼠正常组可见主动脉壁厚薄均匀,内膜完整,无动脉粥样硬化斑块形成,血管中层VSMCs亦未见明显病理变化。20周龄ApoE-/-小鼠模型组主动脉内膜明显增厚,局部可见明显腔内凸起的纤维斑块,纤维帽较完整,内含脂质中心及泡沫细胞,平滑肌细胞、弹力纤维及胶原组织构成纤维帽主要结构;干预组内膜不完整,无连续性,局部内膜明显增厚,并有泡沫细胞聚集。30周龄模型组AS斑块弥漫血管全管腔,纤维肌性成分减少,有坏死中心形成,斑块底部有柳叶状的胆固醇结晶;干预组斑块面积及病变程度较模型组显著减轻,见图1。
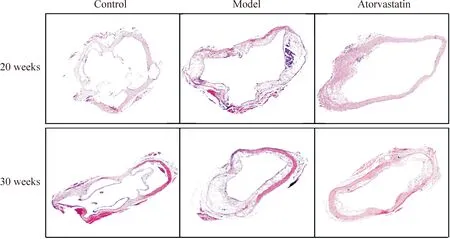
Figure 1.Observation of aortas in each group under optical microscope(HE staining,×40).
3 不同周龄小鼠主动脉粥样硬化斑块面积定量分析
20周龄模型组、阿托伐他汀干预组与正常对照组 AS 斑块面积/动脉总面积分别为0.20±0.03、0.14±0.01与0;30周龄模型组、阿托伐他汀干预组与正常对照组 AS 斑块面积/动脉总面积分别为0.44±0.03、0.23±0.02与0;与20周龄模型组比较,30周龄模型组斑块面积显著增大(P<0.05);而同一周龄小鼠斑块面积相比较,阿托伐他汀干预组显著小于模型组(P<0.05),见图2。
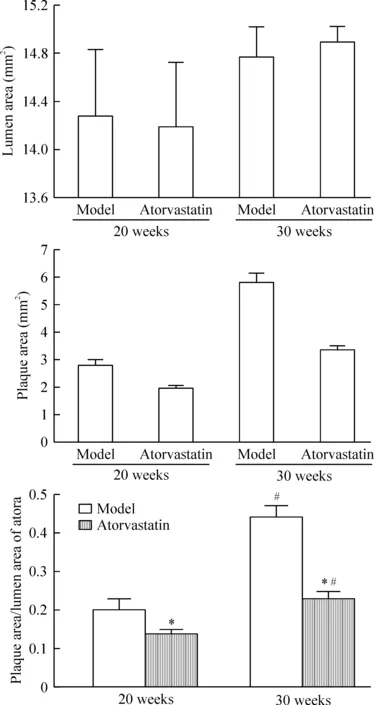
Figure 2.The plaque area/lumen area in different groups. Mean±SD. n=10. *P<0.05 vs model at the same age; #P<0.05 vs 20 weeks in the same group.
4 不同周龄小鼠主动脉TRPC5蛋白免疫组化IA值的表达变化
与正常组C57BL/6J小鼠比较,TRPC5蛋白在ApoE-/-小鼠模型组及阿托伐他汀干预组动脉粥样硬化斑块中的表达明显增高(P<0.01),且以模型组增高最为明显;同周龄ApoE-/-小鼠阿托伐他汀干预组中TRPC5蛋白表达较模型组有所下降(P<0.01)。30周龄干预组与20周龄干预组相比较呈下降趋势(P<0.05)。30 周龄ApoE-/-小鼠模型组与20周龄模型组相比,30周龄C57小鼠与20周龄C57BL/6J小鼠比较均无统计学意义(P>0.05),见图3~4。
5 不同周龄小鼠主动脉TRPC5 mRNA的表达变化
各时点ApoE-/-小鼠模型组的TRPC5 mRNA水平显著高于正常组C57BL/6J小鼠(P<0.05),给药治疗后明显降低(P<0.05)。20周龄正常对照组C57BL/6J小鼠mRNA表达比30周龄正常组略高,但差异无统计学意义(P>0.05)。30周龄时ApoE-/-小鼠模型组TRPC5 mRNA表达稍高于20周龄模型组,但差异无统计学意义(P>0.05);30周干预组较20周干预组相比有降低的趋势,且差异有统计学意义(P<0.05),见图5。
讨 论
ApoE-/-小鼠[6]是目前公认的研究AS发病机制以及抗AS药理学研究等方面最经典的动物模型之一,而C57/BL6J小鼠与其具有相同的遗传背景,因而常在研究中作为健康对照组。ApoE-/-小鼠AS的发病机制是通过影响肝脏对VLDL和乳糜微粒代谢,使胆固醇残粒在血浆堆积引起高脂血症,血脂持续增高,大量胆固醇在血管壁沉积形成AS,这与人类动脉粥样硬化形成过程相似[7]。ApoE-/-小鼠可以自发形成AS,高脂饮食能加速疾病进程。本实验采用高脂饲喂ApoE-/-小鼠建立AS疾病模型,经观察发现随时间延长其血脂持续性增高,20周龄时形成表面的纤维帽,内含脂质、巨噬细胞、VSMCs和泡沫细胞的纤维斑块,此时病变仅局限于部分血管壁;至30周龄演变为粥样斑块,且斑块弥漫整个血管横切面,研究结果与刘剑刚等[8]的研究相一致。
动脉粥样硬化始动环节为血管内皮受损, LDL在内皮下积聚并被自由基氧化,生成氧化修饰的低密度脂蛋白(oxidized LDL, ox-LDL),ox-LDL参与VSMCs增殖与迁移,并促进和加速AS进展[9]。目前已证实VSMCs的增殖和迁移是决定AS及动脉损伤后再狭窄的关键因素[10]。AS发生发展过程中,作为对血管损伤和炎症反应的一部分,VSMCs由收缩表型转变为增殖表型,不断增殖、迁移到内膜,释放大量细胞外基质(extracellular matrix,ECM),并参与纤维斑块的构成。随着AS病变发展,粥样斑块形成并向复合病变进展时,VSMCs发生凋亡并且远大于增殖效应,引起斑块易损甚至破裂。
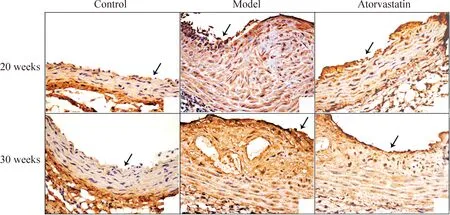
Figure 3.The expression of TRPC5 in the VSMCs of different groups (IHC,×400). The arrows indicate the lumen of the blood vessels.
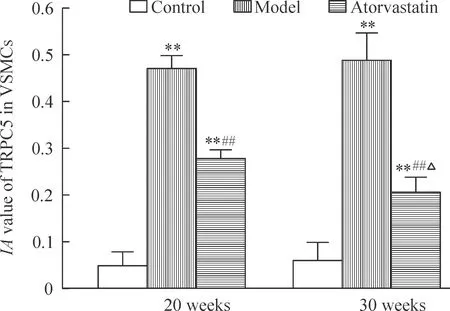
Figure 4.IA value of TRPC5 in the VSMCs of different groups. Mean±SD. n=10. **P<0.01 vs control at the same age; ##P<0.01 vs model at the same age; △P<0.05 vs 20 weeks in the same group.
经研究发现TRPC家族在VSMCs中大量表达,最普遍的是TRPC1、TRPC4和TRPC5[11]。其中TRPC5通道蛋白具有脂质离子亚型受体特性,可识别作为ox-LDL主要成分的溶血磷脂胆碱(lysophosphatidylcholine,LPC),并且被ox-LDL中的1-磷酸鞘氨醇(sphingosine 1-phosphate,S1P)成分激活[12]。本实验观察发现:TRPC5通道蛋白在正常组C57BL/6J小鼠主动脉VSMCs中是稳定低表达的;20周龄ApoE-/-小鼠主动脉VSMCs中表达量明显增高,至30周龄时TRPC5蛋白表达量增加不明显。其机制可能是,高脂血症引起血管管壁ox-LDL明显增高,激活了TRPC5通道,促进VSMCs内Ca2+的增加,并刺激VSMCs由收缩表型转变为增殖表型迁移至血管内膜,吞噬ox-LDL及脂质形成泡沫细胞,参与纤维斑块形成并促进AS进展。随病情进展至粥样斑块时,由于细胞外基质、各种蛋白酶、多种刺激因子及胞内Ca2+超负荷等相互作用,引起细胞损伤和促进VSMCs凋亡程序启动,影响了TRPC5蛋白的表达。
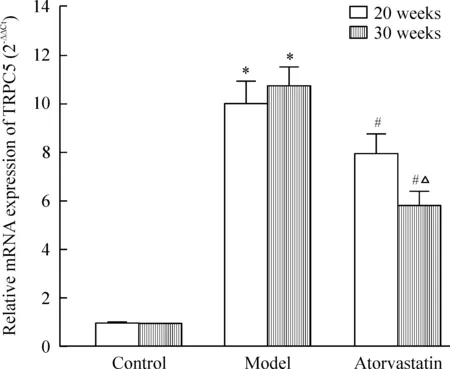
Figure 5.The mRNA expression of TRPC5 in different groups. Mean±SD. n=10. *P<0.05 vs control at the same age; #P<0.05 vs model at the same age; △P<0.05 vs 20 weeks in the same group.
阿托伐他汀属于3-羟基-3-甲基戊二酰辅酶A还原酶抑制剂,许多研究发现他汀类除调脂作用外,还具有改善血管内皮细胞功能、抗炎、抗氧化、抑制VSMCs增生等其它作用[13],并进一步发现他汀可以对多种离子通道进行调控[14]。各种证据亦证明他汀类可以稳定AS斑块[15],对心脑血管发挥保护作用。2013年美国心脏病学会与美国心脏协会联合颁布的最新版降胆固醇治疗降低动脉粥样硬化性心血管疾病(atherosclerotic cardiovascular disease,ASCVD)风险指南[16]中亦强调了他汀类药物在ASCVD一二级预防的重要作用,但目前关于他汀抗AS的具体机制仍不明确。该实验发现阿托伐他汀除了在高脂饲喂的ApoE-/-小鼠中的直接降脂作用外,还可以显著缩小ApoE-/-小鼠主动脉 AS斑块面积并下调TRPC5蛋白的表达,且随治疗时间延长其抑制作用更明显。可能机制为,阿托伐他汀一方面通过加速血液中LDL清除,减少其被氧化为ox-LDL的机会,并减少TRPC5通道的激活从而抑制VSMCs增殖和迁移;另一方面可以通过降低细胞内活性氧,减少氧化应激从而抑制了TRPC5蛋白表达;抑制细胞内钙超载,延缓了动脉粥样硬化的病理进程。
综上所述,我们发现TRPC5在ApoE-/-小鼠AS斑块形成的过程中表达上调,而阿托伐他汀可以下调TRPC5通道表达,且下调作用随时间延长更为显著。因此推测阿托伐他汀可能通过减少TRPC5表达来抑制VSMCs增殖和迁移,从而延缓AS斑块进展。我们将通过后续实验进一步探讨TRPC5在VSMCs增殖与迁移过程中的具体分子机制及阿托伐他汀的作用机制,为AS防治提供新的思路。
[1] Faxon DP, Fuster V, Libby P, et al. Atherosclerotic Vascular Disease Conference:Writing Group III: pathophysio-logy[J]. Circulation, 2004, 109(21): 2617-2625.
[2] Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells[J]. Pharmacol Rev, 2000, 52(4):639-672.
[3] 陈兆煜,袁 乔,谈 智. Caveolae-caveolin-1-PTRF/cavin-1系统与平滑肌细胞迁移:一种可能的机制[J]. 中国病理生理杂志, 2013, 29(5):957-960.
[4] Vazquez G. TRPC channels as prospective targets in atherosclerosis: terra incognita[J]. Front Biosci (Schol Ed), 2012, 4:157-166.
[5] Beech DJ. Canonical transient receptor potential 5[J]. Handb Exp Pharmacol, 2007,179:109-123.
[6] Plump AS, Smith JD, Hayek T, et al. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells[J]. Cell, 1992, 71(2):343-353.
[7] Zhang SH, Reddick RL, Piedrahita JA, et al. Sponta-neous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E[J]. Science, 1992, 258(5081):468-471.
[8] 刘剑刚,董国菊,史大卓,等. 载脂蛋白E基因敲除小鼠不同周龄动脉粥样硬化的病理变化[J]. 中国动脉硬化杂志, 2005, 13(6):689-692.
[9] Massaeli H, Austria JA, Pierce GN. Chronic exposure of smooth muscle cells to minimally oxidized LDL results in depressed inositol 1,4,5-trisphosphate receptor density and Ca2+transients[J]. Circ Res, 1999, 85(6):515-523.
[10]Rudijanto A. The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis[J]. Acta Med Indones, 2007, 39(2):86-93.
[11]Dietrich A, Chubanov V, Kalwa H, et al. Cation channels of the transient receptor potential superfamily: their role in physiological and pathophysiological processes of smooth muscle cells[J]. Pharmacol Ther, 2006, 112(3):744-760.
[12]Xu SZ, Muraki K, Zeng F, et al. A sphingosine-1-phosphate-activated calcium channel controlling vascular smooth muscle cell motility[J]. Circ Res, 2006, 98(11):1381-1389.
[13]Tesfamariam B, Frohlich BH, Gregg RE. Differential effects of pravastatin, simvastatin, and atorvastatin on Ca2+release and vascular reactivity[J]. J Cardiovasc Pharmacol, 1999, 34(1):95-101.
[14]Patel TN, Shishehbor MH, Bhatt DL. A review of high-dose statin therapy: targeting cholesterol and inflammation in atherosclerosis[J]. Eur Heart J, 2007, 28(6):664-672.
[15]林晓燕,林秋平,许昌声,等. 阿托伐他汀通过RXRα介导的抗氧化应激效应抑制高脂喂养糖尿病ApoE-/-小鼠动脉粥样硬化的形成[J]. 中国病理生理杂志, 2014, 30(9):1537-1545.
[16]Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines[J]. J Am Coll Cardiol, 2014,63(25):2889-2934.
Effect of atorvastatin on TRPC5 expression in atherosclerosis of apolipoprotein E-knockout mice
QI Jie1, XU Fang2, MA Hui1, CUI Jian-guo1, ZHANG Qing-tan1
(1DepartmentofGeriatricMedicine,AffiliatedHospitalofBinzhouMedicalUniversity,Binzhou256603,China;2DepartmentofPathophysiology,BinzhouMedicalUniversity,Yantai264003,China.E-mail:qtzhangby@126.com)
AIM: To observe the changes of transient receptor potential channel 5 (TRPC5) in vascular smooth muscle cells (VSMCs) of apolipoprotein E-knockout (ApoE-/-) mice and the effect of atorvastatin interference, and to investigate the mechanism of atorvastatin therapy. METHODS: MaleApoE-/-mice at 6 weeks of age were used to establish the atherosclerosis model by feeding with hyperlipidic diet. The mice were randomly divided into model group and atorvastatin group. The mice in atorvastatin group were lavaged with atorvastatin at 20 mg·kg-1·d-1, while the mice in model group received normal saline. The healthy C57BL/6J mice with the same age and the same genetic background, feeding with ordinary food, served as control group. At the time points of 14 and 24 weeks, the mice were sacrificed. The serum was collected for detecting the lipid levels. The aortic roots of the heart were taken to make paraffin sections with HE staining for measuring and comparing the relative atherosclerotic plaque area in each section. The expression of TRPC5 in VSMCs was examined with immunohistochemical staining. The mRNA levels of TRPC5 in the serum and the thoracoabdominal aorta were measured by real-time PCR. RESULTS: Compared with model group, blood lipids in atorvastatin group were significantly decreased, and the formation of plaque under aorta intima also decreased. The protein expression of TRPC5 in atorvastatin group decreased significantly compared with model group. Compared with 20-week model group, TRPC5 in 30-week model group showed increasing tendency, but has no statistical significance. Compared with 20-week atorvastatin group, TRPC5 of 30-week atorvastatin group declined. CONCLUSION: Atorvastatin suppresses TRPC5 expression, thus attenuating atherosclerotic development inApoE-/-mice.
TRPC5; Apolipoprotein E; Atherosclerosis; Vascular smooth muscle cells; Atorvastatin
1000- 4718(2015)03- 0457- 06
2014- 11- 10
2014- 12- 08
国家自然科学基金资助项目(No. 81401625); 滨州医学院科研启动基金资助项目(No. BY2009KYQD05)
△通讯作者 Tel: 0543-3258527; E-mail: qtzhangby@126.com
R541.4; R972+.6
A
10.3969/j.issn.1000- 4718.2015.03.013
