补肾丸定性定量方法研究
2015-04-07张爱项菲
张爱 项菲
(麻阳县食品药品监督管理局,湖南 麻阳 419400)
补肾丸定性定量方法研究
张爱 项菲
(麻阳县食品药品监督管理局,湖南 麻阳 419400)
目的:建立补肾丸的定性定量标准。方法:用薄层色谱法(TLC)对锁阳、白芍及黄柏进行定性鉴别;用高效液相色谱法(HPLC)测定补肾丸中熟地黄的含量,采用Dikma Diamonsil C18柱(250 mm×4.6 mm,5 μm);流动相:乙腈-0.1%醋酸溶液(16∶84);流速:1.0 mL/min;柱温:35℃;检测波长:334 nm;进样量:20 μL。结果:TLC能明显检出锁阳、白芍及黄柏特征斑点,无阴性药材干扰。毛蕊花糖苷进样量在5.08~81.28 μg范围内线性关系良好,r=0.999 44,平均回收率为98.72%,RSD=0.82%。结论:定性、定量方法简便、准确、重现性好,能有效控制补肾丸的内在质量。
补肾丸;质量标准;薄层色谱法;高效液相色谱法;毛蕊花糖苷
补肾丸是由锁阳、枸杞子、白芍、五味子及熟地黄等10味中药制成的复方制剂,具有锁阳固精,滋阴补肾的功效,广泛应用于肾水不足,阴虚阳亢,头晕咳嗽,腰膝酸痛,梦遗滑精等病症。熟地黄为方中君药,其有效成分为毛蕊花糖苷。其质量标准收载于《中华人民共和国卫生部药品标准中药成方制剂》(第二册),只有显微鉴别,为了保证该药品质量的稳定有效,本研究参考资料文献,对制剂中的锁阳、白芍及黄柏进行定性鉴别,同时用高效液相色谱法(HPLC)对制剂中熟地黄中所含的毛蕊花糖苷进行含量测定。建立了方法可靠、准确,专属性强的质量控制方法,提升了该制剂质量标准。
1 仪器与试药
美国安捷伦 1260高效液相色谱仪(Agilent 1260四元泵,Agilent自动进样器,Agilent数据处理软件系统);CBL9960A超声波清洗器(上海科导超声仪器有限公司);日本岛津AUW220D电子分析天平(十万分之一);毛蕊花糖苷(批号:111530-200706)、熊果酸(批号:110742-200516)、芍药苷(批号:110736-201337)、黄柏对照药材(批号:121510-200703)、盐酸黄柏碱(批号:111895-201202),均购自中国食品药品检定研究院;补肾丸(呼伦贝尔松鹿制药有限公司,6 g/丸,批号:130708,131119,140322,140603);乙腈为色谱纯,甲醇为分析纯,水为超纯水。
2 薄层定性鉴别
2.1 锁阳
取本品6 g,剪碎,加等量硅藻土,研匀,加乙醇 60 mL,超声处理 30 min,滤过,滤液浓缩至1 mL,作为供试品溶液。取缺锁阳的阴性样品,同法制备成阴性对照溶液。再取熊果酸对照品,加甲醇制成每1 mL含0.5 mg的溶液,作为对照品溶液。照薄层色谱法(《中华人民共和国药典》2010年版一部附录 ⅥB)试验,吸取供试品溶液10 μL,对照品溶液4 μL及阴性对照溶液10 μL,分别点于同一硅胶G薄层板上,以甲苯-乙酸乙酯-甲酸(20∶4∶0.5)为展开剂,展开13 cm取出,晾干,喷以10%硫酸乙醇溶液,加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同的紫红色斑点。阴性对照无干扰(图1)。
2.2 白芍

图1 锁阳TLC图
取本品 10 g,剪碎,加等量硅藻土,研匀,加乙醇 60 mL,超声处理 30 min,滤过,滤液蒸干,残渣加水30 mL使溶解,用水饱和的正丁醇振摇提取3次,每次20 mL,合并正丁醇提取液,置水浴上浓缩至1 mL,加适量中性氧化铝,拌匀,干燥,加在中性氧化铝柱(100目,4 g,内径为1 cm),用甲醇50 mL洗脱,收集洗脱液,蒸干,残渣加乙醇1 mL使溶解,作为供试品溶液。取缺白芍的阴性样品,同法制备成阴性对照溶液。再取芍药苷对照品,加乙醇制成1 mL含1 mg的溶液,作为对照品溶液。照薄层色谱法(TLC)(《中华人民共和国药典》2005年版一部附录ⅥB)试验,吸取上述 3种溶液各10 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇(5∶1)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,在100℃加热至斑点显色清晰,供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。阴性对照无干扰(图2)。
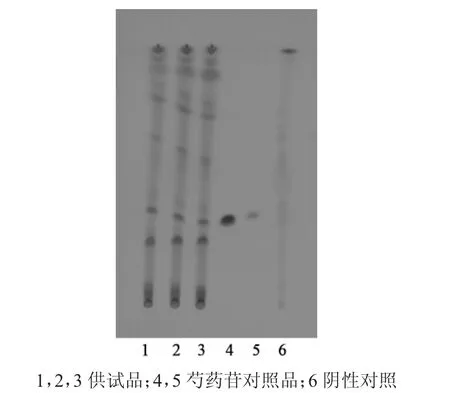
图2 白芍TLC图
2.3 黄柏
取本品6 g,剪碎,加等量硅藻土,研匀,加1%醋酸甲醇溶液50 mL,于60℃超声处理20 min,滤过,滤液浓缩至1 mL,作为供试品溶液。取缺黄柏的阴性样品,同法制备成阴性对照溶液。再取黄柏对照药材0.2 g,加1%醋酸甲醇20 mL,同法制成对照药材溶液。再取盐酸黄柏碱对照品,加甲醇制成每1 mL含 0.5 mg的溶液,作为对照品溶液。照TLC(《中华人民共和国药典》2010年版一部附录ⅥB)试验,吸取上述4种溶液各3~5 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水(30∶15∶4)的下层溶液为展开剂,展开,取出,晾干,喷以稀碘化铋钾试液。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点。阴性对照无干扰(图3)。
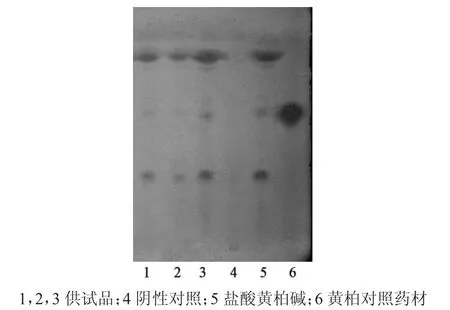
图3 黄柏TLC图
3 熟地黄的含量测定
3.1 供试品溶液的制备
取装量差异项下的本品,剪碎,混匀,取约3 g,精密称定,加入硅藻土3 g,研匀,置具塞锥形瓶中,精密加入甲醇100 mL,密塞,称定质量,加热回流30 min(功率250 W,频率50 kHz),取出,放冷,用甲醇补足减失的质量,摇匀,滤过,取续滤液50 mL,减压回收溶剂至干,残渣用流动相溶解,转移至10 mL量瓶中,加流动相至刻度,摇匀,滤过,取续滤液,即得。
3.2 阴性样品溶液的制备
取除去熟地黄以外的其他药材,按供试品制备方法“3.1”项下方法制成阴性溶液。
3.3 对照品溶液的制备
精密称取干燥至恒重的毛蕊花糖苷对照品10.16 mg,置100 mL量瓶中,加入甲醇使溶解并稀释至刻度,摇匀,作为对照品储备液(浓度为101.6 μg/mL);精密量取上述溶液5 mL,置50 mL量瓶中,加流动相稀释至刻度,即得(浓度为10.16 μg/mL)。
3.4 色谱条件
Dikma Diamonsil C18柱(250 mm×4.6 mm, 5 μm);流动相:乙腈-0.1%醋酸溶液(16∶84);流速:1.0 mL/min;柱温:35℃;检测波长:334 nm;进样量:20 μL。 理论板数按毛蕊花糖苷峰计算应不低于3 000。
3.5 专属性试验
照“3.4”项下色谱条件,分别精密吸取供试品溶液、对照品溶液、阴性样品溶液各10 μL,注入液相色谱仪中,在毛蕊花糖苷对照品色谱相应的位置上,供试品溶液具有相同保留时间的色谱峰,阴性液在此峰位无吸收,对本品中毛蕊花糖苷含量测定无干扰。见图4。
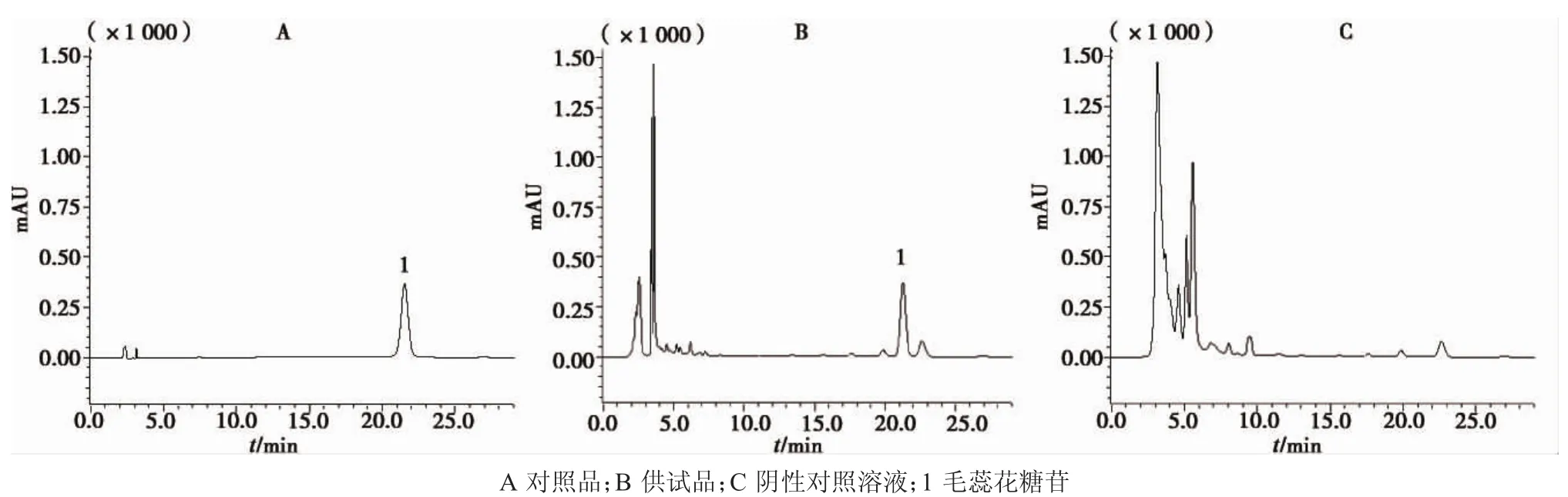
图4 补肾丸HPLC图
3.6 线性范围考察
精密吸取 “3.3”项下浓度为101.6 μg/mL的对照品储备溶液0.5,1.0,2.0,4.0,6.0,8.0 mL至 10 mL量瓶中,加甲醇稀释定容至刻度,按“3.1”项下方法进行测定,以进样量为横坐标X,色谱峰面积为纵坐标Y,计算回归方程,毛蕊花糖苷回归方程为:
Y=3 095.5X-2 799.1。
r=0.999 44,毛蕊花糖苷进样量在5.08~ 81.28 μg/mL范围内线性关系良好。
3.7 精密度试验
精密吸取供试品(批号:140603)溶液10 μL,注入液相色谱仪中,在上述色谱条件下测定,连续进样6次,分别测定各次峰面积,毛蕊花糖苷峰面积的RSD为0.76%。结果表明该色谱系统具有较好的精密度。
3.8 稳定性试验
取供试品(批号:140603)溶液,分别在 0,4, 6,8,12,24 h,按上述色谱条件进样10 μL,测定色谱峰面积,结果毛蕊花糖苷峰面积的RSD为0.91%。表明样品溶液在所考察时间内稳定性良好。
3.9 重现性试验
取同一批供试品(批号:140603),精密称取6份,按供试品测定项下方法操作,重复测定6次,毛蕊花糖苷平均含量为 0.331 0 mg/丸,RSD为 0.75%。表明该方法重现性好。
3.10 加样回收率试验
取已知毛蕊花糖苷含量(批号:140603,毛蕊花糖苷含量0.331 0 mg/丸)的供试品约1.5 g共 9份,分别精密加入浓度为101.6 μg/mL毛蕊花糖苷对照品溶液 0.8,0.8,0.8,1.0,1.0,1.0,1.2,1.2, 1.2 mL,按供试品溶液制备方法操作,测定加标样品中的毛蕊花糖苷含量,计算回收率,结果见表1。
3.11 样品含量测定
取不同批号的样品,按供试品测定项下方法操作,测定毛蕊花糖苷含量,结果见表2。
4 讨论
4.1 经过不断摸索试验,制定了锁阳、白芍及黄柏的鉴别方法,该方法特征斑点明显,色谱分离清晰,可作为补肾丸中锁阳、白芍及黄柏的专属性鉴别。
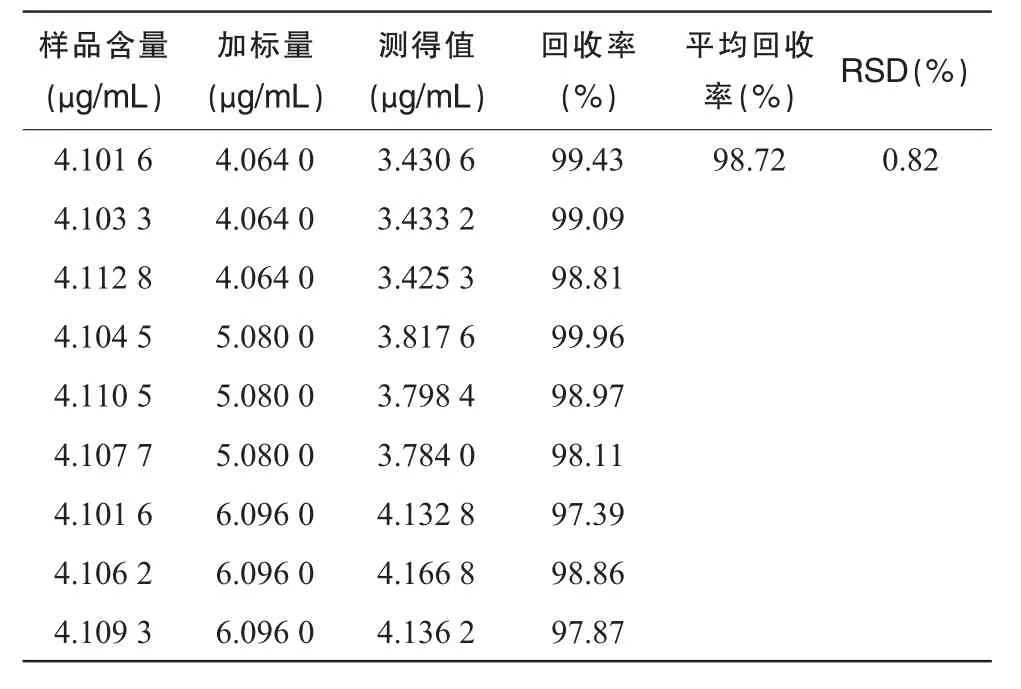
表1 回收试验结果(n=9)
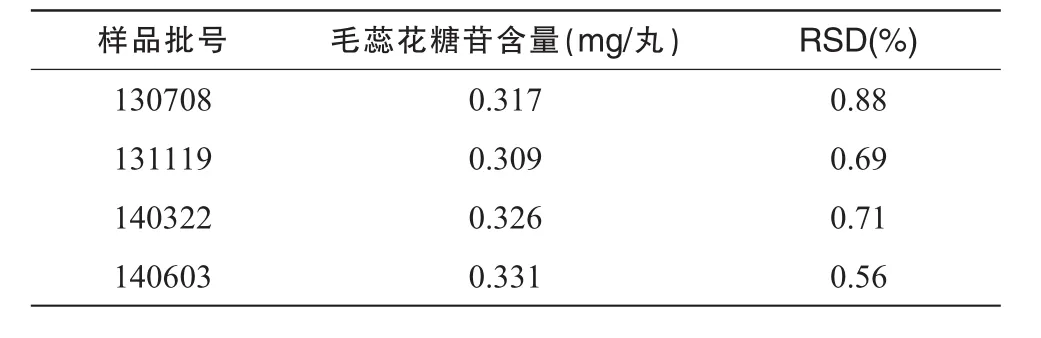
表2 样品含量测定结果
4.2 本试验样品处理方法简单,测定结果准确可靠,可有效控制本制剂的质量,对保证制剂质量有着重要意义。
[1] 国家药典委员会.中华人民共和国药典(一部)[S].北京:中国医药科技出版社,2010.
[2] 中国药品生物制品检定所.中国药品检验标准操作规范[M].北京:中国医药科技出版社,2005:79-95.
[3] 徐灿辉,何维为,何云飞.HPLC法测定六味地黄胶囊中的毛蕊花糖苷、马钱苷、芍药苷和丹皮酚[J].药物评价研究,2014,37(3):257-259.
[4] 李伟,龙书可,刘辉,等.口服清新泡腾片定性定量标准研究[J].中南药学,2010,8(6):410-412.
[5] 陈天朝,瞿来超.HPLC法同时测定地黄中梓醇与毛蕊花糖苷的含量 [J].中国实验方剂学杂志,2011,17(5):105-107.
[6] 宋子荣,谭梓駿,陈百松,等.地黄提取工艺的优化[J].中国实验方剂学杂志,2006,12(1):8.
[7] 陈传福,张培正.两种不同提取熟地黄多糖工艺研究[J].中国食物与营养,2008,8(11):852-854.
[8] 郭晓玲,韩亮,冯毅凡,等.瑶族医药风湿骨痛酒质量标准研究[J].中成药,2007,29(3):386-389.
[9] 宗凯,熊苗苗,谢委,等.胃肠康胶囊质量标准研究[J].医药导报,2010,29(1):92-94.
Study on the Methods for Qualitative and Quantitative Control of Bushen Pills
Zhang Ai,Xiang Fei(Food and Drug Administration Bureau of Mayang County,Hunan Mayang 419400,China)
Objective:To establish the standards for qualitative and quantitative control of Bushen pills.Methods:TLC was used to identify quantitatively Cynomorium songaricum Rupr,Paeoniae radix alba and Phellodendri chinensis cortex.The content of Radix Rehmanniae Praeparata in Bushen pills was determined by HPLC.Dikma Diamonsil column C18(250 mm×4.6 mm,5 μm)was adopted with a mobile phase of acetonitrile-0.1%acetic acid(16∶84)at a flow rate of 1.0 mL/min,the column temperature was at 35℃,the detection wavelength was at 334 nm and the sample size was 20 μL.Results:The feature spots of Cynomorium songaricum Rupr,Paeoniae radix alba and Phellodendri chinensis cortex can be detected by TLC without interference of negative materials.A good linear relationship of acteoside was obtained within the range of 5.08~81.28 μg(r=0.999 44).The average recovery was 98.72%with RSD of 0.82% (n=6).Conclusion:The method is simple,accurate and reproducible and can be used for the effective control of the internal quality of Bushen pills.
Bushen Pills;Quality Standards;TLC;HPLC;Acteoside
10.3969/j.issn.1672-5433.2015.08.004
2015-04-22)
张爱,女,主管药师。研究方向:药物分析。E-mail:569997073@qq.com
项菲,女,主管药师。研究方向:药物分析。通讯作者E-mail:1115844935@qq.com
