Efficient Syntheses of Permethylated Derivatives of Neolamellarin A, a Pyrrolic Marine Natural Product
2015-04-05YINRuijuanJIANGLongWANShengbiaoandJIANGTao
YIN Ruijuan, JIANG Long, WAN Shengbiao, and JIANG Tao
Key Laboratory of Marine Drugs of Ministry of Education of China,School of Pharmacy,Ocean University of China,Qingdao266003,P. R. China
Efficient Syntheses of Permethylated Derivatives of Neolamellarin A, a Pyrrolic Marine Natural Product
YIN Ruijuan, JIANG Long, WAN Shengbiao, and JIANG Tao*
Key Laboratory of Marine Drugs of Ministry of Education of China,School of Pharmacy,Ocean University of China,Qingdao266003,P. R. China
The pyrrole-derived alkaloids with marine origin, especially their permethyl derivatives, have unique structures and promising biological activities. Marine natural product neolamellarins are a collection of lamellarin-like phenolic pyrrole compounds, which can inhibit hypoxia-induced HIF-1 activation. Many pyrrole-derived lamellarin-like alkaloids show potent MDR reversing activity. In this study, five permethylated derivatives of neolamellarin A were synthesized with their MDR reversing activity studied in order to identify new MDR reversal agents. A convergent strategy was adopted to synthesize the permethylated derivatives of neolamellarin A. Pyrrole was first converted into a corresponding N-trisisopropylsilyl (TIPS)-substituted derivative, then through iodination afforded 3,4-diiodinated pyrrole compound. The key intermediate, 3,4-disubstituent-1H-pyrrole, was obtained through desilylation of 3,4-disubstituent-1-TIPS pyrrole , which was prepared from 3,4-diiodinated pyrrole derivative and aryl boronic acid ester through Suzuki cross-coupling reaction between them. Then, the intermediate, 3,4-disubstituent-1H-pyrrole, reacted with fresh phenylacetyl chloride under n-BuLi/THF condition afforded the target compounds. Finally, we obtained five novel pyrrolic compounds, permethylated derivatives of neolamellarin A 16a-e, in 30%-37% yield through five step reactions. The bioactivity testing of these compounds are in process.
neolamellarin A; derivative; synthesis; Suzuki-Miyaura cross-coupling reaction; acylation
1 Introduction
In recent years, the lamellarins and related pyrrole-derived alkaloids as a group of marine alkaloids have attracted more and more attentions due to their promising biological activities (Fanet al., 2008), such as antitumor, multidrug resistance reversal (MDR) and inhibition of HIV-1 integrase and MCV topoisomerase. Liuet al.(2007) separated four new lamellarin-like phenolic pyrroles, neolamellarin A (1), neolamellarin B (2), 5-hydroxyneolamellarin B (3), and 7-hydroxyneolamellarin A (4) from spongeDendrilla nigra(Fig.1). Among these marine natural compounds, 7-hydroxyneolamellarin A shows the most significant anti-tumor activities, for example, inhibiting hypoxia-induced HIF-1 activation (IC501.9 µmol L-1) in T47D cells and vascular endothelial growth factor secretion in breast tumor cells. Neolamellarin A was also found to inhibit hypoxia-inducible factor-1 (HIF-1) activation by 26% at 10 μmol L-1. The difference between neolamellarins and lamellarins is the pattern of oxidation on substituted pyrrole ring. These four new neolamellarins were most similar to lamellarin O (5) in structure (Fig.1).Lamellarin O, which is originally isolated from Australian marine spongeDendrilla cactos(Urbanet al., 1994), exhibits modest cytotoxic activity against both wild-type and MDR tumor cell lines, thus may serve as a new lead in the development of antitumor agents insensitive to MDR (Bogeret al., 1999). Ningalin B (6) (Fig.1) is another lamellarin-like phenolic pyrrole compound, which is isolated from an unidentified Western Australian ascidian of genusDidemnum(Kang and Fenical, 1997). Its permethyl derivative is shown to potently reverse MDR phenotype, desensitizing a resistant human colon cancer cell line (HCT116/VM46) to vinblastine and doxorubicin at lower doses than the prototypical agent verapamil. Permethyl ningalin B (7) (Fig.1) may constitute the initial members of a new class of MDR reversal agents (Bogeret al., 2000). As a result, thede novosynthesis of pyrrole-derived alkaloids with marine origin, especially their permethyl derivatives, have become one of active research fields in recent years (Yinet al., 2014; Menna, 2014).
In 2009, Arafeh and Ullah first synthesized neolamellarin A. They directly builded 3,4-bis(4-methoxyphenyl)-1H-pyrrole core through condensation of vinylogous amide with aminomalonate, then acylation and deprotection of the target compound (Arafeh and Ullah, 2009). Until recently, there is no other report on these four unique structural pyrrolic marine natural products neolamellarinsand their derivatives. Combination of the unique structure of neolamellarin A and pyrrole-derived alkaloids’ promising biological activities, we intend to synthesize a series of permethylated derivatives of neolamellarin A.

Fig.1 The structures of neolamellarin A (1), neolamellarin B (2), 5-hydroxyneolamellarin B (3), 7-hydroxyneolamellarin A (4), lamellarin O (5), ningalin B (6), and permethyl ningalin B (7).
2 Experiments
All starting materials and solvents were obtained from commercial sources and used without further purification. Thin-layer chromatography (TLC) was performed on precoated E. Merck silica-gel 60 F254 plates. Column chromatography was performed on silica gel (200-300 mesh Qingdao China). The synthesis of 3,4-diiodo-1-(triisopropylsilyl)-1H-pyrrole followed the Axford’s report (Axfordet al., 2008).
2.1 Preparation of 4,4,5,5-Tetramethyl-2-Aryl-1,3,2-Dioxaborolane (12a-c)
A mixture of purchased methoxyl subsitituted bromobenzene (10 mmol, 1.87 g), pinacolborane (11 mmol, 2.79 g), KOAc (30 mmol, 2.94 g) and PdCl2(dppf) (300 mg) was heated under nitrogen protection at 80℃ in DMSO (50 mL) for 24 h. The reaction mixture was cooled and then poured into water (500 mL), extracted with CH2Cl2(200 mL, 3 times), then the organic phase washed with water and brine, dried over anhydrous MgSO4, and evaporated. The residue was purified by flash chromatography on silica gel (petroleum ether/ ethyl acetate: 20/1).
2.2 Preparation of 3,4-Bisaryl-1-(Triisopropylsilyl)-1H-Pyrrole (13a-c)
A solution of Na2CO3(30 mmol, 3.18 g) in water (15 mL) was added, in one portion, to a solution of 3,4-diiodo-1-(triisopropylsilyl)-1H-pyrrole 10 (3 mmol, 1.43 g), 4,4,5,5-tetramethyl-2-aryl-1,3,2-dioxaborolane 12a or 12b or 12c (6 mmol, 1.40 g), Pd(PPh3)4(0.3 mmol, 0.36 g) in toluene-methanol (132 mL of a 6:5 mixture). The resulting solution was deoxygenated under reduced pressure and back fi lled with nitrogen. This procedure was repeated five times. The ensuing and magnetically stirred solution was heated at re fl ux for 24 h then cooled to room temperature and diluted with water (450 mL). The resulting mixture was extracted with CHCl3(150 mL, 3 times) and the combined organic phases dried over anhydrous MgSO4, and evaporated. The crude product was purified by flash chromatography on silica gel (petroleum ether/ dichloromethane: 10/1, then 5/1).
2.3 Preparation of 3,4-Bisaryl-1H-Pyrrole (14a-c)
Bu4NF (TBAF) (5.0 mL of a 1.0 mol L-1in THF, 5.0 mmol) was added to a magnetically stirred solution of 3,4-bisaryl-1-(triisopropylsilyl)-1H-pyrrole 13a or 13b or 13c (1.0 mmol, 436 mg) in THF (40 mL). After 20 min the volatile components were removed under reduced pressure and the residue subjected to flash chromatography on silica gel (petroleum ether/ dichloromethane: 2/1, then 1/1).
2.4 Preparation of Acyl Chloride (15a, 15b)
One drop DMF and oxalyl chloride (0.3 mL, 1.8 mmol) was added to a solution of the carboxylic acid (1.2 mmol) in anhydrous CH2Cl2(5 mL) at room temperature under argon atmosphere. After 2 h, the solvent and excess oxalyl chloride were removed, keeping the compound in dry and argon atmosphere till use.
2.5 Preparation of Permethyl Derivatives of Neolamellarin A (16a-e)
N-BuLi (0.6 mL, 1.5 mmol, 2 mol L-1in hexane) was added drop by drop to a solution of 3,4-bisaryl-1H-pyrrole 14a or 14b or 14c (1 mmol) in anhydrous THF (5 mL) at -78℃ under argon atmosphere. After 0.5 h, the fresh acyl chloride 15a or 15b in anhydrous THF (5 mL) was added into the mixture solution. The reaction mixture was allowed to warm up to room temperature and stirred at 35℃ for 10 h. Then the reaction mixture was diluted with saturated aqueous NaHCO3(10 mL), and extracted with EtOAc (10 mL, 3 times). The combined organic layers were washed with brine and dried over anhydrous MgSO4, and evaporated. The residue was purified by flash chromatography on silica gel (petroleum ether/EtOAc: 10/1, then 5/1).
3 Results and Discussion
Methylated neolamellarin A is a pyrrole-derived alkaloid in which 3- and 4-positions of pyrrole ring have an identical substituent group, and N position locates an acylation group. Referring to the synthesis methods of 3,4-bisaryl pyrrole compounds (Yuanet al., 2007; Milgramet al., 2007; Furstneret al., 2002; Banwellet al., 1997; Axfordet al., 2008), there should be two strategies of synthesizing this alkaloid, one is directly building the 3,4-bisaryl-N-acylated pyrrole ring through cyclization reactions from (1Z, 3Z)-1,4-diiodo-1,3-dienes and amides as starting materials or through Piloty-Robinson synthesis from hydrazine and a saturated aldehyde. And the other is to build the 3,4-bisaryl-1H-pyrrole intermediate core through Kumada, Stille or Suzuki cross-coupling reaction, then acylation reaction (Scheme 1).
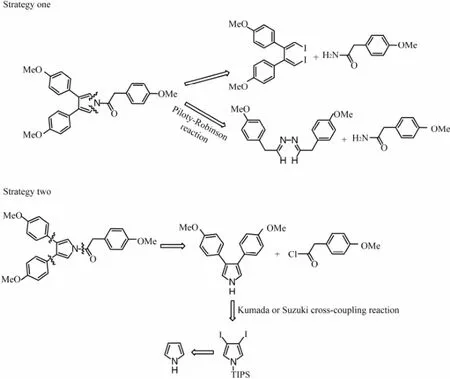
Scheme 1 Retro-synthetic analysis of permethylated neolamellarin A.
In consideration of different substituent groups of 3-, 4-and N positions of pyrrole, a convergent synthesis strategy was used to synthesize the permethylated derivatives of neolamellarin A in this study. We selected pyrrole as the parent compound, and built 3,4-disubstituent-1H-pyrrole through Suzuki cross-coupling reaction first and then acylation at N position.
Several synthetic methods have been developed to construct 3,4-bisaryl-1H-pyrrole (Furstneret al., 2002; Banwellet al., 1997; Axfordet al., 2008; Banwellet al., 2002), and the intermediate 3,4-diiodo-1-(triisopropylsilyl)-1H-pyrrole was proved to be efficient (Axfordet al., 2008; Banwellet al., 2002). We started our synthesis from commercially available pyrrole and bromo anisoles. Firstly, parent pyrrole 8 was converted into corresponding N-trisisopropylsilyl (TIPS)-substituted derivative 9 (96%) (Brayet al., 1990). Using TIPS moiety as a sterically demanding nitrogen substituent to obstruct the attack of electrophilic reagents at the α position on pyrrole ring. Then, two-fold iodination of 9 using molecular iodine in the presence of mercury(ii) acetate afforded the previously reported 3,4-diiodinated pyrrole 10 (72%), which was subjected to react with bis(pinacolato)diboron in the presence of PdCl2(dppf) (Scheme 2). Unfortunately, there was a signi fi cant quantity of the corresponding monoborolated material (22%) accompanying with the 3,4-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-(triisopropylsilyl)-1H-pyrrole 11 (30%).
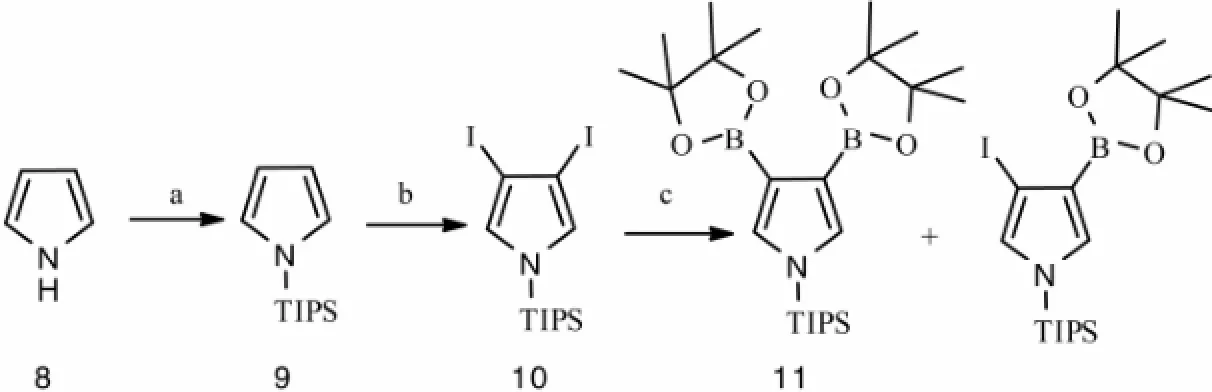
Scheme 2 Synthetic route of compound 11. Reactants and conditions: (a) triisopropylchlorosilane, n-BuLi, THF, -78℃, 2 h; (b) I2, Hg(OAc)2, CH2Cl2, -10℃, 2 h; (c) bis(pinacolato)diboron, PdCl2(dppf), KOAc, DMSO, 80℃, 24 h.
In order to overcome such shortage, we used 3,4-diiodopyrrole derivative 10 and an easily preparing aryl boronic acid ester 12a-c to constructing the C-C bond through Suzuki-Miyaura cross-coupling reaction. Fortunately, the principal product was 3,4-bisaryl pyrrole, and mono-ary- lated pyrrole was not found in products even when short reaction times and 1:1 stoichiometries were tried (Scheme 3 and Table 1).
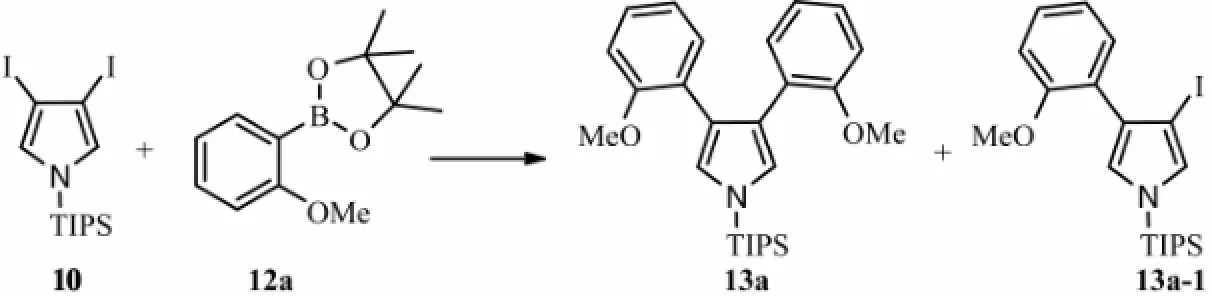
Scheme 3 Suzuki-Miyaura cross-coupling reaction of compound 10 and 12a.

Table 1 Yields of 13a and 13a-1 under different reaction conditions
Under the optimal condition, compounds 13a-c were synthesized in 37.4%-42.0% yield (Scheme 4). After desilylation under Bu4NF condition (Brayet al., 1990), the key intermediates, 3,4-bisaryl-1H-pyrrole 14a-c, were yielded in an excellent range from 90% to 99%.
The acylated reaction of 3,4-bisaryl-1H-pyrrole 14c with purchased phenylacetyl chloride failed under NaH/DMF and n-BuLi/THF conditions. In consideration of severe condition of this acylation reaction, we decided to synthesize fresh acyl chloride. The 4-methoxypheny- lacetic acid was synthesized through witting and oxidation reaction ofp-anisaldehyde (Liet al., 2011; He and Yudin, 2011). The reaction of carboxylic acids with oxalyl chloride in anhydrous CH2Cl2with DMF as the catalyst gave the expected acyl chloride 15a, 15b. Then using the fresh acyl chloride 15a, 15b and 3,4-bisaryl-1H-pyrrole 14a-c as reactants under n-BuLi/THF condition, target products 16a-e were yielded in moderate yields (Scheme 5).
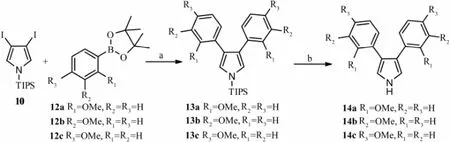
Scheme 4 Synthetic route of compound 14a-c. Reactants and conditions: (a) Pd(PPh3)4, Na2CO3, PhMe:MeOH (6:5), 65℃, 24 h; (b) Bu4NF, THF, 25℃, 25 min.
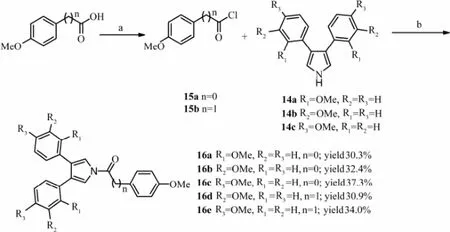
Scheme 5 Synthetic route of compound 16a-e. Reactants and conditions: (a) oxalyl chloride, DMF (one drop), anhydrous CH2Cl2, r.t., 2 h; (b) n-BuLi THF, -78℃ then 35℃, 10 h.
In this study, we adopted the convergent strategy to synthesize neolamellarin A derivatives. Through Suzuki-Miyaura cross-coupling and acylation reactions, we modified the 3,4- and N-positions of pyrrole, and gained5 neolamellarin A derivatives. The bioactivity of these compounds will be determined.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21171154 and 91129706) and Special Fund for Marine Scientific Research in the Public Interest (01005024).
Appendix
The compounds 10, 12a-c, 13a-c, 14a-c, 16a-e were characterized by means of melting point, nuclear magnetic resonance and high-resolution mass spectroscopy. Melting points were determined on a Mitamura-Riken micro-hot stage without correction.1H NMR and13C NMR spectra were obtained on a Bruker 600 spectrometer with tetramethylsilane (Me4Si) as internal standard, and chemical shifts were recorded in δ values. Mass spectra were recorded on a Q-TOF Globalmass spectrometer.
3,4-diiodo-1-(triisopropylsilyl)-1H-pyrrole (10) white solid, yield 80.6%; mp 74-77℃ (ref. mp 75-78℃).
2-(2-methoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxa borolane (12a) White solid 1.99 g, yield 85.1%; mp 75-77℃;1H NMR (600 MHz, CDCl3) δ 7.68-7.66 (dd,J=7.1, 1.7 Hz, 1H), 7.40-7.37 (m, 1H), 6.94-6.92 (t,J=7.4 Hz, 1H), 6.85-6.84 (d,J=8.3 Hz, 1H), 3.82 (s, 3H), 1.34 (s, 12H).
2-(3-methoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxa borolane (12b) Sticky liquid 1.78 g, yield 76.1%;1H NMR (600 MHz, CDCl3) δ 7.42-7.41 (d,J=7.1 Hz, 1H), 7.34 (d,J=2.8 Hz, 1H), 7.32-7.29 (t,J=7.1 Hz, 1H), 7.03-7.01 (m, 1H), 3.84 (s, 3H), 1.35 (s, 12H)..
2-(4-methoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxa borolane (12c) Sticky liquid 2.05 g, yield 87.6%;1H NMR (600 MHz, CDCl3): δ 7.78-7.76 (d,J=8.3 Hz, 2H), 6.91-6.89 (d,J=8.5 Hz, 2H), 3.81 (s, 3H), 1.35 (s, 12H).
3,4-bis(2-methoxyphenyl)-1-(triisopropylsilyl)-1H-p yrrole (13a) White solid 0.53 g, yield 40.5%; mp 114-115℃;1H NMR (600 MHz, CDCl3) δ 7.20-7.17 (t,J=7.9 Hz, 2H), 6.92-6.91 (d,J=7.7 Hz, 2H), 6.87 (m, 4H), 6.76-6.73 (m, 2H), 3.70 (s, 6H), 1.54-1.46 (m, 3H), 1.17-1.16 (d,J=7.5 Hz, 18H);13C NMR (151 MHz, CDCl3) δ 159.26 (2C), 137.52 (2CH), 128.94 (2CH), 125.32 (2C), 123.74 (2C), 121.00 (CH), 113.79 (CH), 111.28 (CH), 55.05 (2CH3), 17.86 (6CH3), 11.61 (3CH); HRMS (ESI)m/z: calcd for M+C27H38NO2Si, 436.2666; found, M+436.26669.
3,4-bis(3-methoxyphenyl)-1-(triisopropylsilyl)-1H-p yrrole (13b) White solid 0.49 g, yield 37.4%; mp 150-151℃;1H NMR (600 MHz, CDCl3) δ 7.14-7.12 (m, 4H), 7.03-6.98 (m, 4H), 6.84 (d,J=5.8 Hz, 2H), 3.50 (s, 6H), 1.50 (m, 3H), 1.18 (s, 9H), 1.16 (s, 9H);13C NMR (151 MHz, CDCl3) δ 156.44 (2C), 130.55 (2C), 128.58 (2CH), 126.42 (2C), 124.42 (2CH), 120.30 (2CH), 111.44 (2CH), 111.03 (2CH), 55.30 (2CH3), 17.93 (6CH3), 11.67 (3CH); HRMS (ESI)m/z: calcd for M+C27H38NO2Si, 436.2666; found, M+436.26665.
3,4-bis(4-methoxyphenyl)-1-(triisopropylsilyl)-1H-p yrrole (13c) White solid 0.55 g, yield 42.0%; mp 135-137℃;1H NMR (600 MHz, CDCl3) δ 7.24-7.22 (d,J=8.6 Hz, 4H), 6.84-6.82 (d,J=8.6 Hz, 4H), 6.76 (s, 2H), 3.67 (s, 6H), 1.54-1.44 (m, 3H), 1.18 (s, 9H), 1.16 (s, 9H).
3,4-bis(2-methoxyphenyl)-1H-pyrrole (14a) White solid 0.41 g, yield 93.2%; mp 148-150℃;1H NMR (600 MHz, CDCl3) δ 8.33 (s, NH), 7.15-7.17 (m, 4H), 7.00-7.01 (d,J=2.76, 2H), 6.82-6.86 (m, 4H), 3.50 (s, 6H);13C NMR (151 MHz, CDCl3) δ 156.44 (2C), 130.63 (2CH), 126.81 (2CH), 125.91 (2C), 120.16 (2CH), 120.07 (2C), 117.90 (2CH), 110.65 (2CH), 54.98 (CH3), 54.95 (CH3); HRMS (ESI)m/z: calcd for M+C18H18O2N, 280.1332; found, M+280.1334.
3,4-bis(3-methoxyphenyl)-1H-pyrrole (14b) Sticky liquid 0.38 g, yield 86.4%;1H NMR (600 MHz, CDCl3) δ 8.48 (s, NH), 7.21-7.18 (t,J=7.9 Hz, 2H), 6.92-6.90 (m, 4H), 6.86 (s, 2H), 6.77-6.75 (dd,J=8.2, 2.0 Hz, 2H), 3.70 (s, 6H);13C NMR (151 MHz, CDCl3) δ 159.26 (2C), 137.06 (2C), 129.03 (2CH), 123.36 (2C), 121.10 (2CH), 117.43 (2CH), 113.79 (2CH), 111.58 (2CH), 55.01 (2CH3); HRMS (ESI)m/z: calcd for M+C18H18O2N, 280.1332; found, M+280.1335.
3,4-bis(4-methoxyphenyl)-1H-pyrrole (14c) White solid 0.42 g, yield 95.5%; mp 100-104℃;1H NMR (600 MHz, CDCl3) δ 8.26 (s, NH), 7.22-7.20 (d,J=8.6 Hz, 4H), 6.85-6.82 (m, 6H), 3.81 (s, 6H);13C NMR (151 MHz, CDCl3) δ 157.74 (2C), 129.55 (4CH), 128.33 (2C), 123.00 (2C), 116.72 (2CH), 113.54 (4CH), 55.17 (2CH3); HRMS (ESI)m/z: calcd for M+C18H18O2N, 280.1332; found, M+280.1338.
(3,4-bis(2-methoxyphenyl)-1H-pyrrol-1-yl)(4-metho xyphenyl)methanone (16a) White solid 125mg, yield 30.3%; mp 145-146℃;1H NMR (600 MHz, CDCl3) δ 7.88-7.85 (d,J=8.8 Hz, 2H), 7.52 (s, 2H), 7.22-7.19 (m, 2H), 7.17-7.16 (dd,J=7.1, 1.6 Hz, 2H), 7.02-7.00 (d,J= 9.3 Hz, 2H), 6.88-6.86 (t,J=7.2 Hz, 2H), 6.83-6.82 (d,J= 7.7 Hz, 2H), 3.90 (s, 3H, OCH3), 3.46 (s, 6H, 2OCH3);13C NMR (151 MHz, CDCl3) δ 166.74 (C), 162.81 (C), 156.50 (2C), 131.95 (2CH), 130.30 (2CH), 127.98 (2CH), 125.34 (C), 125.26 (2C), 124.32 (2C), 120.41 (2CH), 120.27 (2CH), 113.75 (2CH), 110.75 (2CH), 55.52 (CH3), 55.00 (2CH3); HRMS (ESI)m/z: calcd for M+C26H24NO4, 414.1700; found, M+414.1701.
(3,4-bis(3-methoxyphenyl)-1H-pyrrol-1-yl)(4-metho xyphenyl)methanone (16b) White solid 134 mg, yield 32.4%; mp 132-134℃;1H NMR (600 MHz, CDCl3), δ 7.86-7.84 (d,J=8.6 Hz, 2H), 7.43 (s, 2H), 7.22-7.19 (t,J=7.7 Hz, 2H), 7.04-7.02 (d,J=8.6 Hz, 2H), 6.89-6.87 (d,J=7.9 Hz, 2H), 6.84-6.80 (m, 4H), 3.92 (s, 3H, OCH3), 3.70 (s, 6H, 2OCH3);13C NMR (151 MHz, CDCl3) δ 166.62 (C), 162.89 (C), 159.14 (2C), 135.09 (2C), 131.78 (2CH), 129.01 (2CH), 127.58 (2C), 124.51 (C), 120.86 (2CH), 119.67 (2CH), 113.71 (2CH), 113.63 (2CH), 112.45 (2CH), 55.31 (CH3), 54.86 (2CH3); HRMS (ESI)m/z: calcd for M+C26H24NO4, 414.1700; found, M+414.1702.
(3,4-bis(4-methoxyphenyl)-1H-pyrrol-1-yl)(4-metho xyphenyl)methanone (16c) White solid 1154 mg, yield37.3%; mp 120-121℃;1H NMR (600 MHz, CDCl3) δ 7.84-7.83(d,J=8.8 Hz, 2H), 7.35 (s, 2H), 7.20-7.18 (d,J=8.8 Hz, 4H), 7.02-7.00 (d,J=8.8 Hz, 2H), 6.84-6.82 (d,J=8.8 Hz, 4H), 3.90(s, 3H), 3.80(s, 6H);13C NMR (151 MHz, CDCl3) δ 166.80 (C), 162.85 (C), 158.62 (2C), 131.94 (2CH), 129.58 (4CH), 127.73 (C), 126.40 (2C), 124.82 (2C), 119.28 (2CH), 113.86 (2CH), 113.69 (4CH), 55.48 (CH3), 55.19 (2CH3); HRMS (ESI)m/z: calcd for M+C26H24NO4, 414.1700; found, M+414.1704.
1-(3,4-bis(3-methoxyphenyl)-1H-pyrrol-1-yl)-2-(4-m ethoxyphenyl)ethanone (16d) White solid 132 mg, yield 30.9%; mp 107-109℃;1H NMR (600 MHz, CDCl3) δ 7.49 (s, 2H), 7.28-7.27 (d,J=7.7 Hz, 2H), 7.23-7.20 (t,J=7.9 Hz, 2H), 6.94-6.92 (d,J=8.5 Hz, 2H), 6.87-6.81 (m, 6H), 4.16 (s, 2H), 3.83 (s, 3H), 3.71 (s, 6H);13C NMR (151 MHz, CDCl3) δ 168.40 (C), 159.35 (2C), 158.94 (C), 135.09 (2C), 130.21 (2CH), 129.25 (2CH), 128.31 (C), 124.82 (2C), 121.03 (2CH), 117.86 (2CH), 114.35 (2CH), 113.82 (2CH), 112.80 (2CH), 55.27 (CH3), 55.09 (2CH3), 40.51 (CH2); HRMS (ESI)m/z: calcd for M+C27H26NO4, 428.1856; found, M+428.1859.
1-(3,4-bis(4-methoxyphenyl)-1H-pyrrol-1-yl)-2-(4-m ethoxyphenyl)ethanone (16e) White solid 145 mg, yield 34.0%; mp 107-109℃;1H NMR (600 MHz, CDCl3) δ 7.41 (s, 2H), 7.28-7.26 (d,J=8.4 Hz, 2H), 7.18-7.16 (d,J=8.3 Hz, 4H), 6.93-6.91 (d,J=8.4 Hz, 2H), 6.85-6.84 (d,J=8.3 Hz, 4H), 4.14 (s, 2H), 3.82 (s, 9H);13C NMR (151 MHz, CDCl3) δ 168.33 (C), 158.88 (C), 158.62 (2C), 130.47 (C), 130.20 (2CH), 129.60 (4CH), 126.30 (2C), 117.17 (2C), 114.31 (2CH), 114.09 (2CH), 113.71 (4CH), 55.19 (3CH3), 40.49 (CH2); HRMS (ESI)m/z: calcd for M+C27H26NO4, 428.1856; found, M+428.1860.
Arafeh, K. M., and Ullah, N., 2009. Synthesis of neolamellarin A, an inhibitor of hypoxia-inducible factor-1.Natural Product Communication, 4 (7): 925-926.
Axford, L. C., Holden, K. E., Hasse, K., Banwell, M. G., Steglich, W., Wagler, J., and Willis, A. C., 2008. Attempts to mimic key bond-forming events associated with the proposed biogenesis of the pentacyclic lamellarins.Australian Journal of Chemistry, 61 (2): 80-93.
Banwell, M. G., Bray, A. M., Edwards, A. J., and Wong, D. J., 2002. Rapid and convergent assembly of the polycyclic framework assigned to the cytotoxic marine alkaloid halitulin.Journal of the Chemical Society,Perkin Transactions 1, 1 (11): 1340-1343.
Banwell, M. G., Flynn, B. L., Hamel, E., and Hockless, D. C. R., 1997. Convergent syntheses of the pyrrolic marine natural products lamellarin-O,lamellarin-Q, lukianol-A and some more highly oxygenated congeners.Chemical Communications, 1 (2): 207-208.
Boger, D. L., Boyce, C. W., Labroli, M. A., Sehon, C. A., and Jin, Q., 1999. Total syntheses of ningalin A, lamellarin O, lukianol A, and permethyl storniamide A utilizing heterocyclic azadiene Diels-Alder reactions.Journal of the American Chemical Society, 121 (1): 54-62.
Boger, D. L., Soenen, D. R., Boyce, C. W., Hedrick, M. P., and Jin, Q., 2000. Total synthesis of ningalin B utilizing a heterocyclic azadiene Diels-Alder reaction and discovery of a new class of potent multidrug resistant (MDR) reversal agents.Journal of Organic Chemistry, 65 (8): 2479-2483.
Bray, B. L., Mathies, P. H., Naef, R., Solas, D. R., Tidwell, T. T., and Artis, D. R., 1990. N-(Triisopropylsily1) pyrrole. A progenitor ‘par excellence’ of 3-substituted pyrroles.Journal of Organic Chemistry, 55 (28): 6317-6328.
Fan, H., Peng, J., Hamann, M. T., and Hu, J. F., 2008. Lamellarins and related pyrrole-derived alkaloids from marine organisms.Chemical. Reviews, 108 (1): 264-287.
Furstner, A., Krause, H., and Thiel, O. R., 2002. Efficient relay syntheses and assessment of the DNA-cleaving properties of pyrrole alkaloid derivatives permethyl sterniamide A, lycogalic acid A dimethyl ester, and the halitulin core.Tetrahedron, 58 (32): 6373-6380.
He, Z., and Yudin, A. K., 2011. Amphoteric α-boryl aldehydes.Journal of the American Chemical Society, 133 (35): 13770-13773.
Kang, H., and Fenical, W., 1997. Ningalins A-D: Novel aromatic alkaloids from a Western Australian ascidian of the genusDidemnum. Journal of Organic Chemistry, 62 (10): 3254-3262.
Li, Q., Jiang, J., Fan, A., Cui, Y., and Jia, Y., 2011. Total synthesis of lamellarins D, H, and R and ningalin B.Organic Letter, 13 (2): 312-315.
Liu, R., Liu, Y., Zhou, Y. D., and Nagle, D. G., 2007. Molecular-targeted antitumor agents. 15. Neolamellarins from the marine spongeDendrilla nigrainhibit hypoxia-inducible factor-1 activation and secreted vascular endothelial growth factor production in breast tumor cells.Journal of Natural Products, 70 (11): 1741-1745.
Menna, M., 2014. Important classes of bioactive alkaloids from marine ascidians: Structures, isolation and bioactivity.Current Topics in Medicinal Chemistry, 14 (2): 207-223.
Milgram, B. C., Eskildsen, K., Richter, S. M., Scheidt, W. R., and Scheidt, K. A., 2007. Microwave-assisted piloty-robinson synthesis of 3,4-disubstituted pyrroles.Journal of Organic Chemistry, 72 (10): 3941-3944.
Urban, S., Butler, M. S., and Capon, R. J., 1994. Lamellarins O and P: New aromatic metabolites from the Australian marine spongeDendrilla cactos.Australian Journal of Chemistry, 47 (10): 1919-1924.
Yin, R. J., Jiang, L., Wan, S. B., and Jiang, T., 2014. Recent progress in the research on lamellarin o and related pyrrole-derived alkaloids from marine organisms.Zhongguo Haiyang Yaowu, 33 (1): 75-82.
Yuan, X., Xu, X., Zhou, X., Yuan, J., Mai, L., and Li, Y., 2007. Copper-catalyzed double N-alkenylation of amides: An efficient synthesis of di- or trisubstituted N-acylpyrroles.Journal of Organic Chemistry, 72 (4): 1510-1513.
(Edited by Qiu Yantao)
(Received April 24, 2013; revised May 15, 2013; accepted January 4, 2015)
© Ocean University of China, Science Press and Spring-Verlag Berlin Heidelberg 2015
* Corresponding author. Tel: 0086-532-82032712 E-mail: jiangtao@ouc.edu.cn
杂志排行

Journal of Ocean University of China的其它文章
- Impacts of the Two Types of El Niño on Pacific Tropical Cyclone Activity
- Numerical Simulation of Typhoon Muifa (2011) Using a Coupled Ocean-Atmosphere-Wave-Sediment Transport (COAWST) Modeling System
- Estimating the Turbulence Characteristics in the Bottom Boundary Layer of Monterey Canyon
- Composition and Origin of Ferromanganese Crusts from Equatorial Western Pacific Seamounts
- Hydroelastic Analysis of a Very Large Floating Structure Edged with a Pair of Submerged Horizontal Plates
- A Storm Surge Intensity Classification Based on Extreme Water Level and Concomitant Wave Height