氩粒子系统的分子动力学模拟
2015-04-01张丽娟
张丽娟
(晋中学院信息技术与工程学院,山西晋中030619)
(编辑 郭继荣)
0 引言
分子动力学模拟方法(molecular dynamicssimulation,MDS)是一种计算机模拟实验的方法,是当前人们研究凝聚态系统的重要工具,原子的运动轨迹和原子运动过程中的各种微观细节都可以通过这种方法得到,它对理论计算和实验起到了有力的补充作用.这种方法是按体系内每个粒子所遵从的动力学规律即牛顿运动规律来建立粒子的运动方程组,通过解方程组可以得到系统中每个粒子在每个时刻的坐标和动量,也就是能确定粒子的轨迹,根据经典力学和统计物理理论,已知粒子的坐标和动量我们可以用统计计算的方法得到系统的各种性质,比如:系统的各种热力学性质、粒子的均方位移、关联函数等.所以分子动力学模拟能够让我们看到系统在一段时间内的发展过程[2].
1 模拟的基本原理
对我们要研究的系统首先要选取作用势的模型,对于一个粒子间的相互作用势能是球对称的多粒子体系,它的哈密顿量可以写为:

其中,rij是第i个粒子和第j个粒子之间的距离.微正则系综受到的约束有四个:能量守恒、体积不变、粒子数不变、动量恒等于零.系统中粒子的运动方程组可以从哈密顿量推出,如下所示:

采用有限差分法将微分方程化为有限差分方程来求解该方程组,数值求解该方程组,我们需要将此方程组的求解变成求解以下方程组:

从以上方程组可以得出,t+h时刻的坐标位置可以从前面t和t-h时刻这两步的空间坐标位置及t时刻的作用力计算得到.若令:

则式(3)可以写成如下形式:
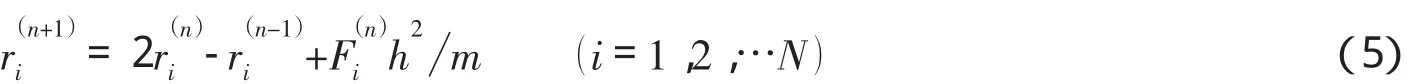

需要注意的是,在第n+1步算出的速度是前一时刻,即第n步的速度,因而动能的计算比势能的计算落后一步.
2 模拟的步骤设计
根据上面的原理我们设计的微正则系综(粒子数恒定、体积恒定、能量恒定)的分子动力学模拟步骤如下:
(4)计算第n步的速度
在上面的算法中,动能的计算比势能的计算要落后一步,而且这种算法不是自启动的,只有给出初始位置}和空间位置}才能求出微分方程组的解,

按照改进后的算法模拟的步骤如下:
(5)重复进行第(2)步
3 平衡化的方法
大多数情况下,精确的初始条件是很难得到的,那么我们可以先给出一个合理的初始条件,接下来在模拟的时候不断调节系统的能量使它达到给定的值.其方法为:首先求解系统的运动方程组,从中解出若干步的结果,接下来根据解出的结果计算体系的能量,看能量是否守恒而且等于我们给定的值,如若不然,那么我们就需要通过调整速度来使能量守恒,也就是用一个标度因子乘以速度,标度因子一般取为[2]:

然后回到第一步,对下一时刻的运动方程求解.反复进行上面的过程,直到系统达到平衡.
采用对速度标度的办法,可以使速度发生很大变化[2],为了消除速度标度带来的后果,模拟过程中必需要有足够的时间让体系再次建立平衡,在到达趋衡阶段以后,必须检验粒子的速度分布是否符合麦克斯韦-玻尔兹曼分布[5].
4 计算结果
计算的时候取256个氩原子作为我们的模拟系统,粒子间的相互作用势为雷纳德-琼斯势[6]:

其中,-ε是两分子处于稳定平衡状态时的势能值,也即势能的最小值,当r=216σ 时,U(r )=-ε;σ 是两分子间作用势为零时它们的间距,即当r=σ时,U(r)=0.该体系的粒子限制在一个立方体的箱子中,为了使密度守恒我们采用周期性边界条件,边界上采用最小像力约定.我们采用自然单位制,长度和时间的标度单位分别为 σ 和 (m σ248ε )12,对氩原子的时间单位为3×10-12s,这样就使得运动方程为无量纲形式.我们研究相图上的一点 (T*, ρ*)= (0.722,0.831 34),立方体箱子的尺寸为6.75.初始条件定为:系统中每一个粒子处于面心立方格子的格点上,而速度按相应温度下的玻尔兹曼分布抽样取值,为了比较势能的不同截断对模拟结果的影响,我们取rc=2.5和rc=3.6,速度标度因子为:

反复上面的速度调节,直到系统能量达到给定值,下面是计算结果:
4.1 能量演化过程
以下是256个氩原子系统在T*=0.722,ρ*=0.83134,rc=2.5时,动能,势能和总能量的演化过程.
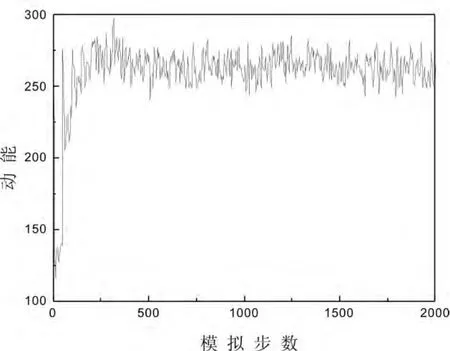
图1 动能演化图

图2 势能演化图

图3 总能量演化图
从上面各图可以看出,微正则系综的分子动力学模拟中,总能量是恒定的,从总能量的演化过程图可以明显地看出模拟的平衡化过程,其平衡化过程是通过对粒子速度的调节跳跃式地达到的.动能和势能不是守恒量,但几百步之后它们能给出持续的确定的平均值.
4.2 轨迹分析
我们在计算中初始条件令各个原子位于一个面心立方格子的格点上,模拟中随着时间的推移系统内原子的分布逐渐趋于无序,当原子分布完全处于无序状态时,也即体系达到稳定状态,这时我们就可以计算体系的各个物理量从而研究体系的各种性质.图4是体系内原子的分布随时间的变化图,从图中可以看出,初始状态体系的原子分布处于有序状态,模拟10步之后体系的有序状态逐渐被打破,至到最后体系内原子的分布完全处于无序状态.

图4 各个时期256个氩原子的位置
4.3 径向分布函数
径向分布函数是一个用来研究物质的有序性的参数,指给定某个粒子的坐标,其他粒子在空间的分布几率,是反映物体结构的特征物理量[2].分子动力学计算径向分布函数的方法为[7,8]:
其中,N为分子数目,T为计算的时间步数,δr为设定的距离差,ΔN为介于r→r+dr间的分子数目.图5是温度为 (T*, ρ*)= (0.722,0.83134 )和 (T*, ρ*)= (2.53,0.636 )时氩原子体系的径向分布函数,从 (T*, ρ*)=(0.722,0.831 34)图中可以看出,当r>3.5σ时径向分布函数的值趋近于1,当r<0.89σ时径向分布函数的值为零.这说明在我们所计算的氩粒子系统中,粒子之间的最小距离为0.89σ,而且平均看来,当粒子与粒子的距离超过3.5σ时,体系中粒子的分布是均匀的.在r~1.1σ处径向分布函数有最大值,这就说明体系中在距离粒子为1.1σ的地方附近其他粒子出现的几率是最大的,此处的局域密度应该是最大的.曲线中其他峰值虽比第一个峰值要小,但是径向分布函数在此处有极大值,说明此处粒子分布的局域密度要大于平均密度.径向分布函数反映出体系中原子的聚集特性,可借此了解体系的结构[2].
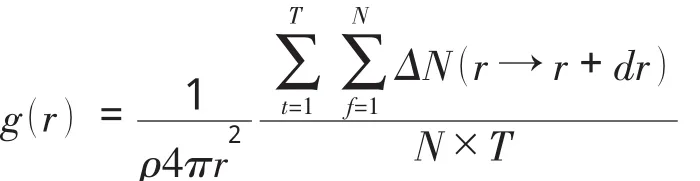
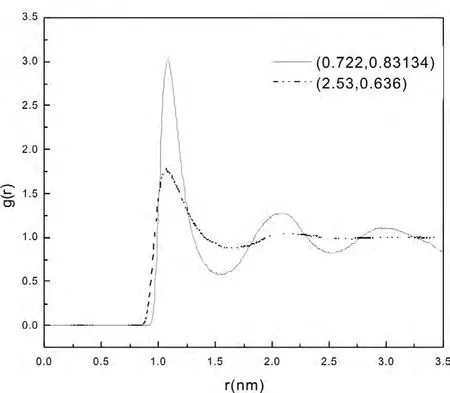
图5 不同温度下的径向分布函数
从两曲线的对比可以看出,(T*, ρ*)= (2.53,0.636 )时的第一峰值要比 (T*, ρ*)= (0.722,0.831 34)时的低.这说明:体系的配位数随着温度的升高而降低,有序度降低,这是符合热力学规律的.(T*, ρ*)=(2.53,0.636 )时的曲线比 (T*, ρ*)= (0.722,0.831 34)时的曲线有一向r减小方向的平移,原因是:温度高的原子动能比较大能够克服它们之间的排斥作用,原子之间可以靠得更近.两条曲线的共同特点是:随着r的增大,体系的各个峰值逐渐减小,说明氩粒子系统短程有序、长程无序的特征.
5 结论
分子动力学模拟方法的应用已经取得了许多重要的成果,可以广泛地用于物理、化学、生物、材料、医学等各个领域.本文通过对256个氩粒子系统进行微正则系综分子动力学模拟得出了系统的能量演化过程、粒子的轨迹、系统的径向分布函数,对了解体系特征有重要意义.
[1]陈正隆,徐为人,汤立达.分子模拟的理论与实践[M].北京:化学工业出版社,2007.
[2]Frenkel&Smit.分子模拟—从算法到应用[M].汪文川,译.北京:化学工业出版社,2002.
[3]D.W.Heemann.理论物理学中的计算机模拟方法[M].秦克诚,译.北京:北京大学出版社,1996.
[4]马文淦.计算物理学[M].北京:科学出版社,2005.
[5]汪志诚.热力学统计物理[M].北京:高等教育出版社,2003.
[6]毋志民,何焕典,罗强,等.银、钴和铂原子纳米团簇熔化过程的分子动力学模拟[J].原子与分子物理学报,2004(51):213~215.
[7]Bixon M,Jortner J.Energetic and thermodynamics sizeeffectsin molecular clusters[J].J.Chem.Phys.,1989,91(3):1631~1642.
[8]Schmidt M,Kusche R,et al.Negativeheat capacity for aclusterof 147 sodiumatoms[J].Phys.Rev.Lett.,2001,86(7):1191~1194.
