Cr/Ti掺杂石墨烯活化CO2分解的理论研究
2015-03-23施龙献杨转霞范美玲王罗新
施龙献, 杨转霞, 范美玲, 王罗新
(武汉纺织大学材料科学与工程学院, 武汉 430200)
Cr/Ti掺杂石墨烯活化CO2分解的理论研究
施龙献, 杨转霞, 范美玲, 王罗新
(武汉纺织大学材料科学与工程学院, 武汉 430200)
为筛选更高效的CO2分解还原的催化材料, 采用密度泛函理论中的B3LYP方法结合6-31G(d)基组, 研究了Cr、 Ti掺杂石墨烯对CO2分子的吸附与催化分解的机理. 研究结果表明: CO2分子在Cr元素和Ti元素掺杂石墨烯表面的吸附为放热过程, 吸附能分别为28.3 kcal/mol和7.7 kcal/mol, 两种元素掺杂石墨烯催化CO2分子中C-O断裂的活化能分别为15.5 kcal/mol和7.8 kcal/mol, 反应过程遵循插入-消除原理, 计算结果为CO2还原的催化剂设计提供理论指导.
掺杂石墨烯; 二氧化碳; 分解; 密度泛函理论
1 引 言
导致全球变暖的温室气体CO2的资源化利用一直是人们的关注焦点[1], 其中, 将CO2转化为其他化学品是其资源化利用的有效途径之一. 人们陆续以CO2为原料制备出了甲烷、 碳氢化合物、 甲醇、 二甲醚、 高级醇、 甲酸、 甲酸盐、 甲酰胺等化合物[2,3]. 相关的研究目前依然活跃, Schmeier等[4]利用水溶性的Ir(III)氢化物为催化剂, 将CO2氢化还原. Motokura等[5]通过具有均相催化功能的高活性、 高选择性的铜-膦络合物实现了CO2的硅氢加成, 得到了烷基硅甲酯; Huang等[6]通过镍钳型化合物催化转化CO2制得了甲醇衍生物; Angamuthu等[7]通过铜配合物电催化CO2得到了草酸盐; Tsutsumi等[8]通过双功能有机小分子催化剂活化CO2与环氧化合物反应, 生成了环状碳酸酯; Bhavani等[9]和Sarusi等[10]通过CO2和甲烷的反应制得了CO和H2. CO2在室温下比较稳定, 在以上所述的过程中,CO2的活化起着非常关键的作用.
CO2的高效活化一直是人们关注的重点, 总体来说, 活化CO2的机理主要是利用物质表面(催化剂)的氧空缺、 缺陷、 以及表面与CO2的电子转移等来实现CO2的活化. 目前也已经有很多涉及CO2活化的实验和理论研究. 通过理论计算, 对于 Fe(111)[11]、 Fe(110)[12]、 Co(110)[12]、 Ni(110)[12]、 Cu(110)[12]、 B[13]、 W-C(0001)[14]、 W-C-Co合金[14]、 CeO2(110)[15]、 TiO2(110)[16,17]等表面活化CO2的机理已有一定认识, 对于筛选合适的CO2分解还原催化材料具有重要指导意义.
近年来, 石墨烯作为催化剂的载体已经引起了人们广泛的关注. 石墨烯是一种单层碳原子紧密相邻的蜂窝晶格结构, 拥有卓越的催化性能[18,19]. 当Al、 Ga、 Si、 N、 B原子掺杂时表现出特别的性质[20-23]. 本研究中, 我们通过理论计算对Cr、 Ti掺杂石墨烯活化CO2的作用和机理进行了探索, 以期获得更高效的CO2分解还原催化材料.
2 计算模型与方法
为简化结构, 我们选用锯齿型三角石墨烯为计算的结构模型, 其每一边拥有5个结构单元的长度(5×5×5), 由氢原子封端使其饱和. 图1给出了原型石墨烯、 Cr掺杂石墨烯(Cr-graphene)和Ti掺杂石墨烯(Ti-graphene)的结构. 所有计算通过Gaussian 03程序包完成[24], 具体采用密度泛函理论(DFT)B3LYP方法结合6-31G(d)基组进行计算. 优化的基态和过渡态结构分别通过频率和IRC计算确认.
3 结果与讨论
经优化后的石墨烯、 Cr掺杂石墨烯(Cr-graphene)和Ti掺杂石墨烯(Ti-graphene)结构如图1所示, 从图中可以看出, Cr、 Ti原子掺杂后, Cr和Ti原子不能和石墨烯的碳原子处于同一平面, 石墨烯中的C-C为1.424 Å, 掺杂后的Cr-C和Ti-C键长增长, 分别为1.836 Å和1.926 Å.

图1 优化的石墨烯(graphene)、 Cr掺杂石墨烯(Cr-graphene)和Ti掺杂石墨烯结构(Ti-graphene)Fig. 1 The optimized geometries of graphene, Cr-doped graphene (Cr-graphene) and Ti-doped graphene (Ti-graphene)
3.1 CO2在Cr-graphene表面的吸附和分解
要催化活化CO2, 首先要求CO2能够吸附在催化剂表面. 我们首先考察石墨烯是否对CO2具有催化作用, 我们发现, CO2与石墨烯并不能形成稳定的复合物结构, 即CO2并不能很好吸附在石墨烯表面, 表明单纯的石墨烯对CO2不具有催化效应.
考察CO2在Cr-graphene表面的吸附情况, 图2给出了优化得到的CO2吸附在Cr-graphene表面形成复合物的结构, 记为Rco2@Cr-graphene. 从优化的结构中可以看出, CO2与Cr-graphene发生作用后, CO2中的双键与Cr产生络合, 导致CO2分子发生变形, 其中OCO键角由180°变为141.5°, 且分子中的C=O键变长. 我们对络合物中的C-O1键断裂进行扫描, 发现随着C-O1键伸长, 体系存在过渡态, 通过进一步优化得到了C-O1键断裂的过渡态结构, 记为TSco2@Cr-graphene, 如图2所示. 频率计算表明此过渡态存在唯一虚频为424.4 cm-1, 对应于结构中的C-O1键的伸缩振动. 通过内禀反应路径(IRC)计算进一步确认了过渡态, 如图3所示. 过渡态中CO2分子进一步靠近Cr-graphene结构, 这可从二氧化碳分子中C、 O1原子与Cr原子的距离变化看出, 表明CO2分子与Cr-graphene相互作用程度加强.
对比Rco2@Cr-graphene和TSco2@Cr-graphene结构可以发现, 络合物向过渡态演化过程中, 随着C-O1键长由1.273 Å增长为1.670 Å, 此时C-O1键活化并发生断裂, CCrO1键角增大了16.8°, Cr-O1键长缩短了0.249 Å. 表明过渡态结构形成时, Cr-graphene结构中的Cr与O1原子间电子作用增强, 形成了Cr-O1键. Cr与二氧化碳中的C原子距离缩短了0.075 Å, 表明Cr-C键形成. 同时, Cr-C1键断裂, Cr与C1的距离增长至2.018 Å.
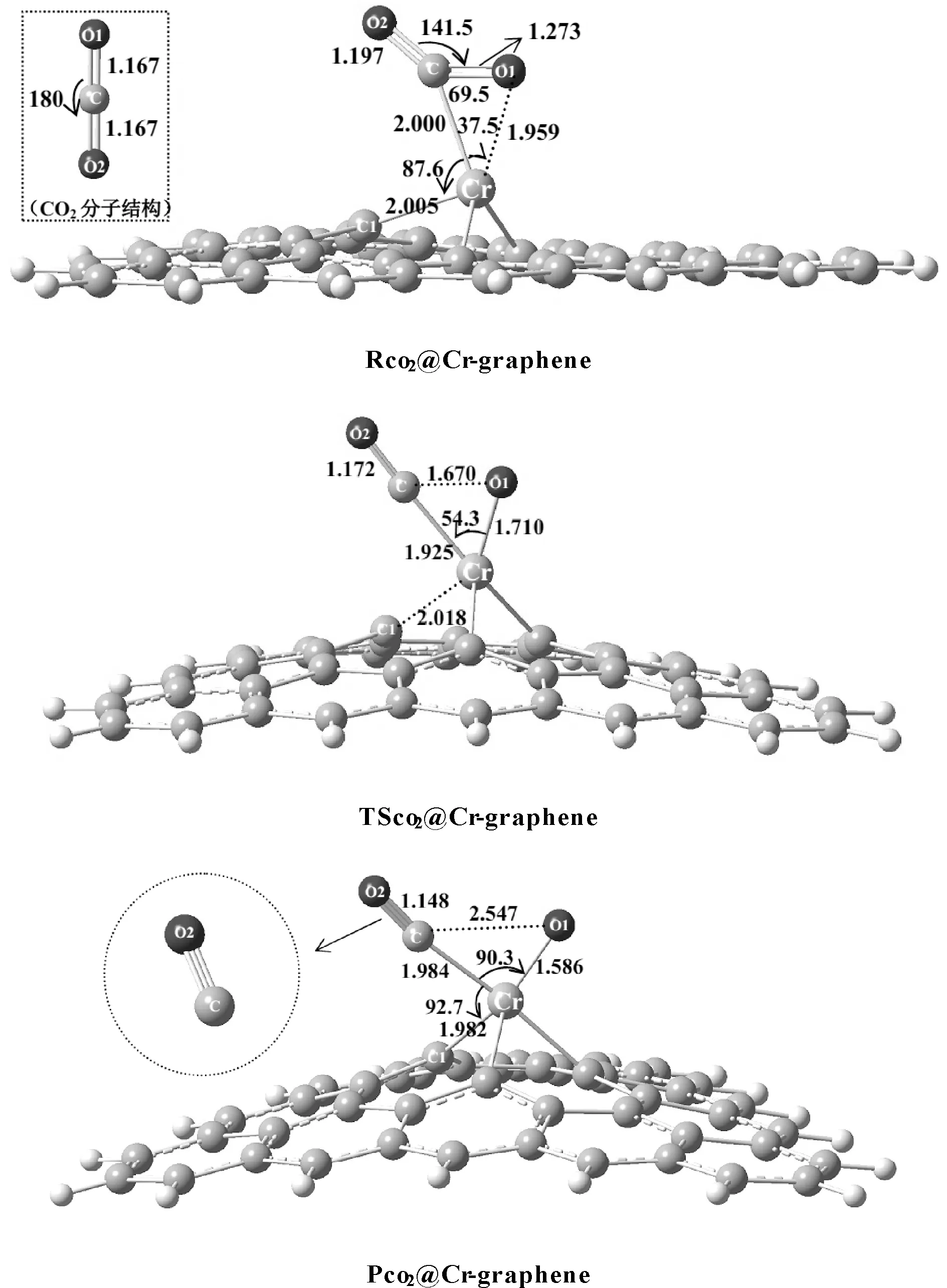
图2 CO2在Cr-graphene表面的吸附形成的复合物Rco2@Cr-graphene、 CO2分解的过渡态TSco2@Cr-graphene和分解中间产物Pco2@Cr-grapheneFig. 2 The optimized complex of CO2 adsorbed on the surface of Cr-graphene (Rco2@Cr-graphene), the transition state of CO2 decomposition(TSco2@Cr-graphene) and the intermediate of CO2 decomposition (Pco2@Cr-graphene)
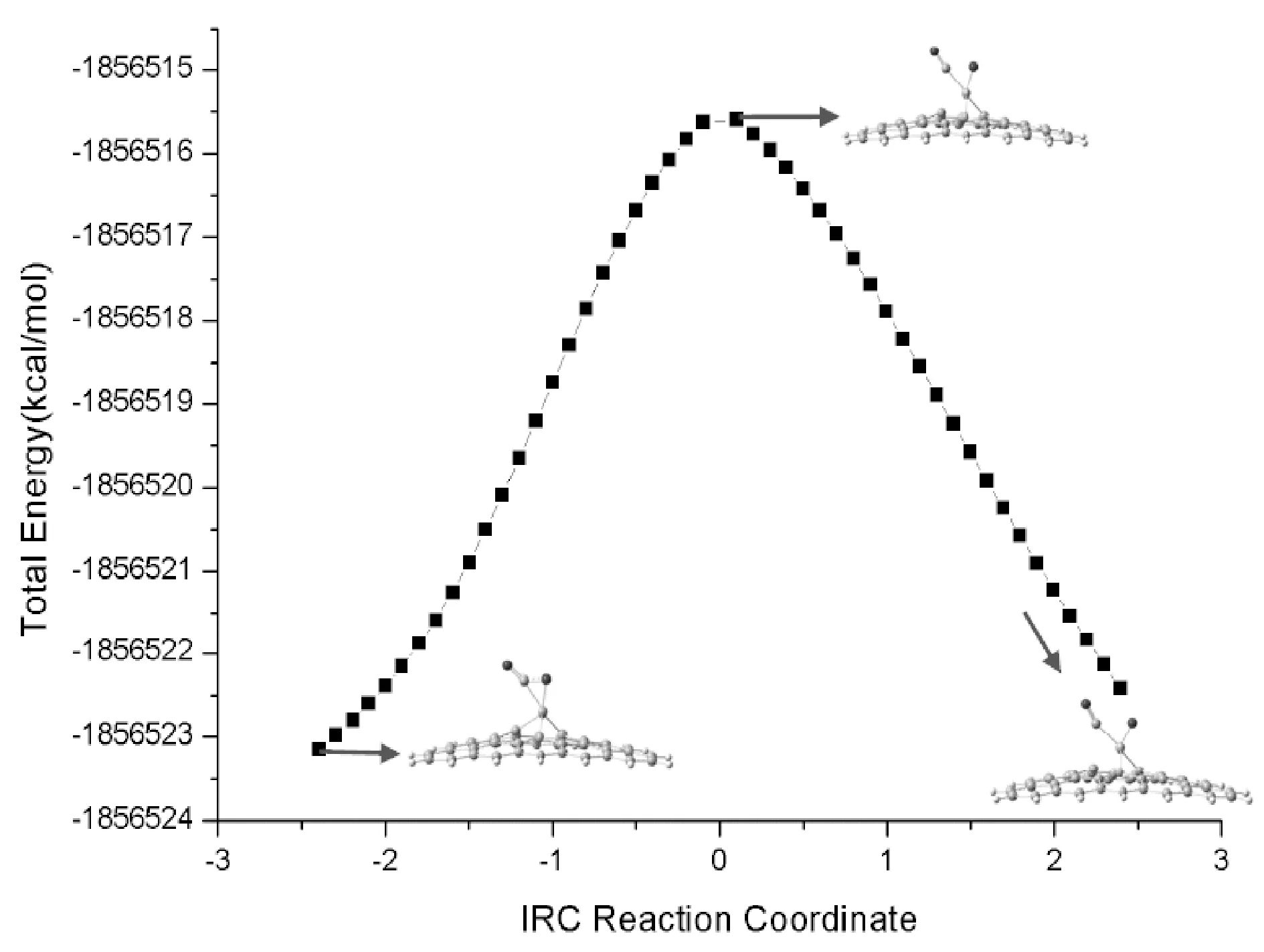
图3 CO2在Cr-graphene表面分解的内禀反应路径(IRC)分析Fig. 3 Intrinsic reaction path (IRC) analysis of the decomposition of CO2 on the surface of Cr-graphene
随着C-O1键进一步伸长,最终生成CO2解离的中间产物Pco2@Cr-graphene,如图2所示.可以看出,这一中间解离产物结构中Cr原子已经插入到C-O1键中,Cr-O1键继续缩短,CCrO1键角继续增大,Cr-C键则增长0.0590 Å,Cr-C1重新形成.
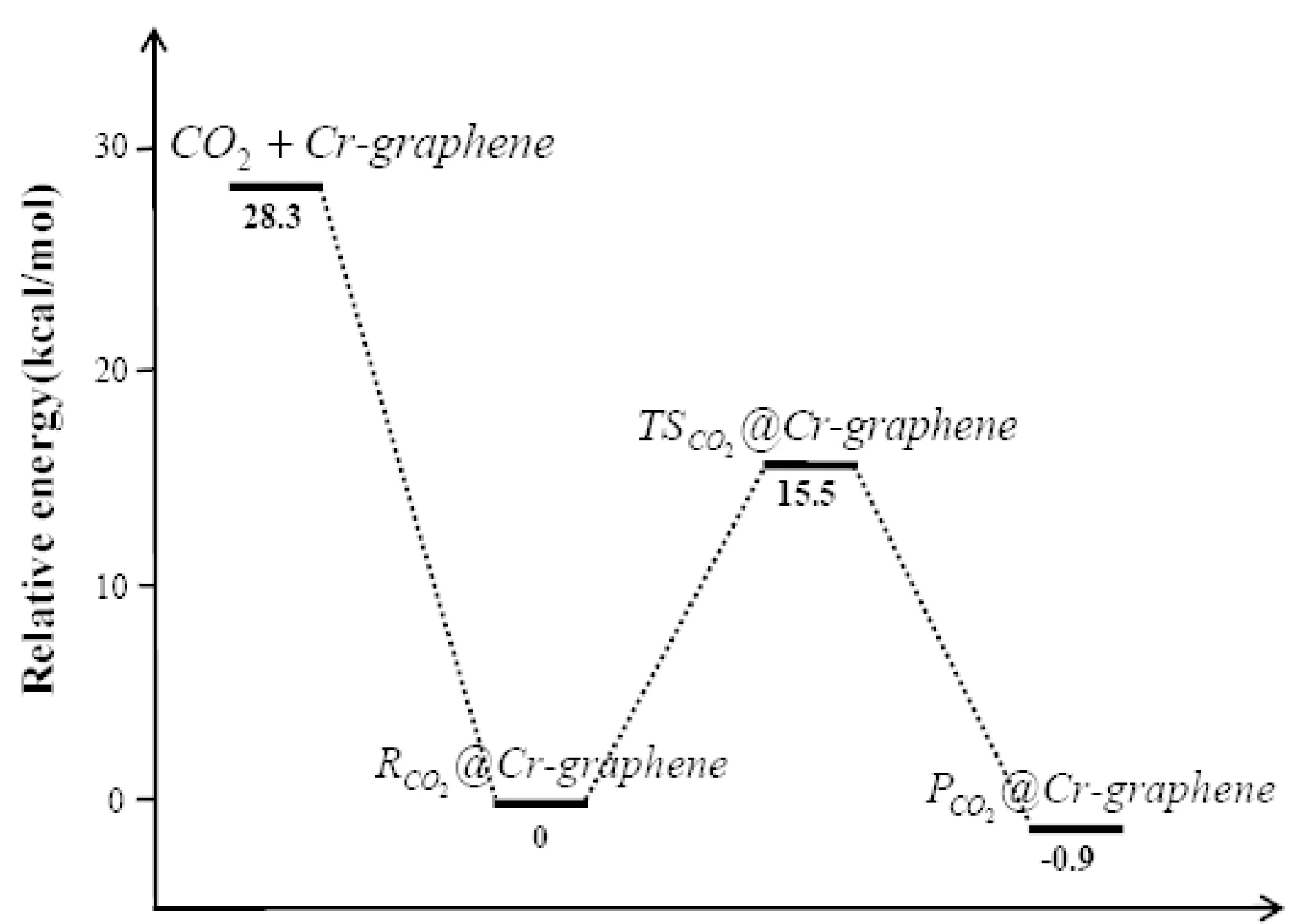
表1给出了CO2分子在不同元素掺杂石墨烯表面发生解离的相关结构的能量.从能量数据分析来看,CO2分子吸附于Cr-graphene表面形成络合物,其能量降低了28.3 kcal/mol,表明Cr-graphene表面吸附CO2分子在能量上是有利的,吸附为自发过程.吸附后发生CO2分子中的C-O键断裂所需要克服的能垒为15.5 kcal/mol,最终形成的解离中间产物比吸附形成的络合物能量低0.89 kcal/mol,因此,CO2的整个吸附和分解的过程为一放热过程.Cr掺杂石墨烯催化分解CO2为CO和O-Cr-graphene,反应的机理同Mo、Mo+、Nb、Nb+催化分解CO2的相同[25,26],是一个插入-消除的反应过程.
表1 CO2分子在不同元素掺杂石墨烯表面吸附和分解的相关结构的能量
Table 1 Energies of the related structures formed by CO2adsorbed to the surfaces of the doped graphene

SpeciesTotalEnergywithZPEcorrection(a.u.)Relativeenergy(kcal/mol)Adsorptionenergy(kcal/mol)CO2-188.57074--Cr-graphene-2769.47261--Ti-graphene-2574.55704--RCO2@Cr-graphene-2958.088530-28.3aTSCO2@Cr-graphene-2958.0637715.5a-PCO2@Cr-graphene-2958.08995-0.89a-RCO2@Ti-graphene-2763.140050-7.70bTSCO2@Ti-graphene-2763.127567.84b-PCO2@Ti-graphene-2763.134803.30b-
a: relative to the energy of RCO2@Cr-graphene
b: relative to the energy of RCO2@Ti-graphene
3.2 CO2在Ti-graphene表面的吸附和分解
采用相同的计算方法, 我们对CO2分子在Ti元素掺杂石墨烯表面发生吸附和催化分解的过程进行了研究. 图4给出了CO2在Ti-graphene表面的吸附形成的复合物Rco2@Ti-graphene、 CO2分解的过渡态TSco2@Ti-graphene和分解中间产物Pco2@ Ti-graphene. 和Cr掺杂石墨烯吸附催化CO2分解过程类似, Ti掺杂石墨烯吸附CO2分子后, 导致CO2结构变形, 使得其中与Ti发生络合作用的C=O1双键活化. 进一步发生CO1键解离形成的过渡态虚频为307.0 cm-1, IRC分析对这一过渡态结构进行了确认(图5所示). 从络合物到过渡态, 再到中间产物, Ti掺杂石墨烯催化CO2的结构变化与Cr掺杂体系基本类似.
同样, CO2分子在Ti元素掺杂石墨烯表面吸附是一个放热过程, 吸附后体系能量降低7.7 kcal/mol(表1所示), 过渡态TSco2@ Ti-graphene和分解中间产物Pco2@ Ti-graphene的能量相对于Rco2@Ti-graphene分别为7.8 kcal/mol和3.3 kcal/mol. 整个反应过程依然为一放热过程, 遵循了插入-消除反应机理.

图4 CO2在Ti-graphene表面的吸附形成的复合物Rco2@Ti-graphene、 CO2分解的过渡态TSco2@ Ti-graphene和分解中间产物Pco2@ Ti-grapheneFig. 4 The optimized complex of CO2 adsorbed on the surface of Ti-graphene (Rco2@Ti-graphene), the transition state of CO2 decomposition(TSco2@Ti-graphene) and the intermediate of CO2 decomposition (Pco2@Ti-graphene)
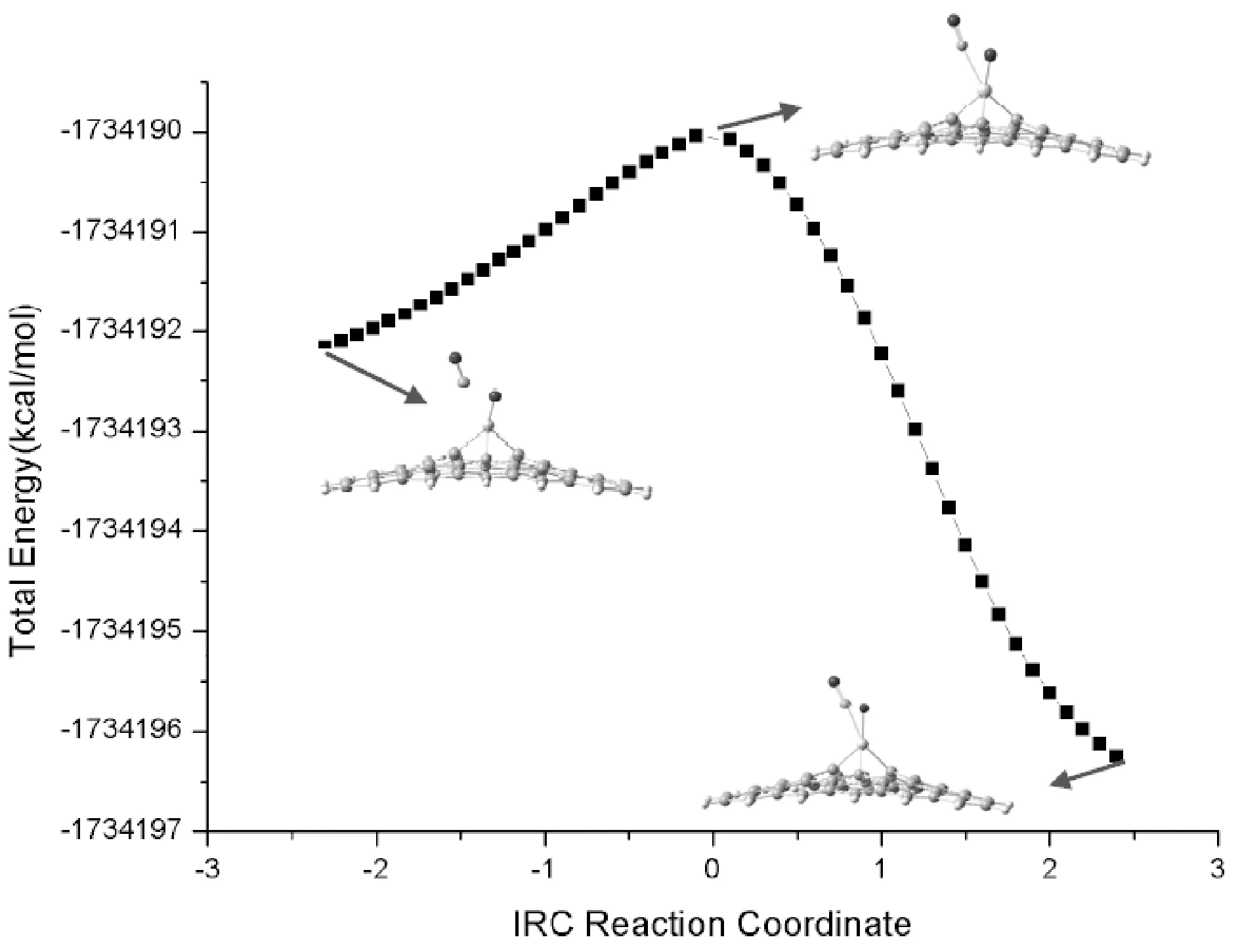
图5 CO2在Ti-graphene表面分解的内禀反应路径(IRC)分析Fig. 5 Intrinsic reaction path (IRC) analysis of the decomposition of CO2 on the surface of Ti-graphene
3.3 与其他催化分解CO2体系的比较
图6给出了CO2分子在Cr-graphene和Ti-graphene表面发生分解反应的势能面, 为便于比较, 我们总结了近年来文献报道的部分过渡金属及氧化物催化剂催化分解CO2的活化能垒, 如表2所示. 可看出, Cr-graphene 和Ti-graphene 表现出了极好的催化性能, 其催化CO2分解的活化能垒比Cu、 Fe、 CeO2催化剂还低, 接近于Ni和Co金属催化剂. 值得注意的是, Ti掺杂石墨烯表现出了最好的催化性能, CO2分解的活化能垒只有7.8 kcal/mol, 因此, 制备Ti掺杂的石墨烯, 有望用于CO2高效还原的候选催化剂.
表2 不同催化剂催化分解CO2的活化能垒
Table 2 Activation energy barriers of the decomposition of CO2catalyzed by different catalysts

CatalystEnergyBarrier(kcal/mol)CatalystEnergyBarrier(kcal/mol)Fe(111)21.7[11]Cu(111)48.4[27]Fe(110)27.1[12]Cu(211)44.3[27]Co(110)12.6[12]Cu(643)46.4[27]Ni(110)12.2[12]Cr-graphene15.5Cu(110)39.5[12]Ti-graphene7.80CeO2(110)85.8[15]
4 结 论
采用DFT(B3LYP/6-31G(d))方法, 我们研究了CO2分别与Cr、 Ti掺杂石墨烯表面的吸附与分解反应机理. 计算结果表明, Cr和Ti元素掺杂的石墨烯能有效吸附CO2分子, 同时, 两种元素掺杂的石墨烯催化CO2分子的解离反应遵循插入-消除机理, 与某些过渡金属原子及氧化物相比, Cr和Ti掺杂石墨烯对CO2的催化分解表现出了更低的活化能垒.
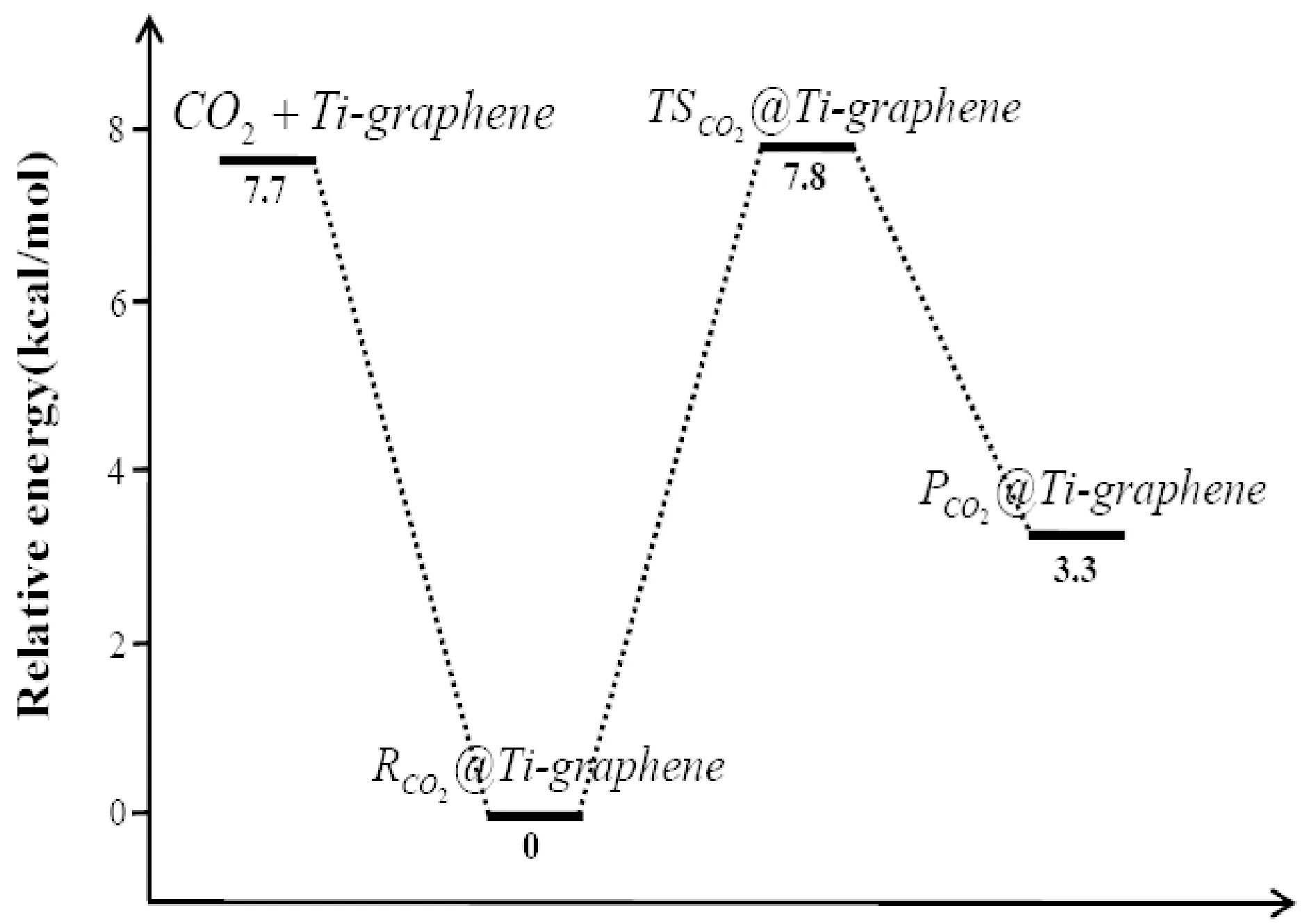
图6 CO2分子在Cr-graphene和Ti-graphene表面催化分解的势能面Fig. 6 Potential energy surfaces for the decomposition of CO2 catalyzed by the Cr-graphene and Ti-graphene
[1] Cao L, Bala G, Caldeira K,etal. Importance of carbon dioxide physiological forcing to future climate change [J].Proc.Natl.Acad.Sci.USA, 2010, 107(21): 9513.
[2] Wang W, Wang S P, Ma X B,etal. Recent advances in catalytic hydrogenation of carbon dioxide [J].Chem.Soc.Rev., 2011, 40(7): 3703.
[3] Centi G, Quadrelli E A, Perathoner S. Catalysis for CO2conversion: a key technology for rapid introduction of renewable energy in the value chain of chemical industries [J].EnergyEnviron.Sci., 2013, 6(6): 1711.
[4] Schmeier T J, Dobereiner G E, Crabtree R H,etal. Secondary coordination sphere interactions facilitate the insertion step in an iridium(III) CO2reduction catalyst [J].J.Am.Chem.Soc., 2011, 133(24): 9274.
[5] Motokura K, Kashiwame D, Takahashi N,etal. Highly active and selective catalysis of copper diphosphine complexes for the transformation of carbon dioxide into silyl formate [J].Chem.Eur.J., 2013, 19(30): 10030.
[6] Huang F, Zhang C G, Jiang J L,etal. How does the nickel pincer complex catalyze the conversion of CO2to a methanol derivative? A computational mechanistic study [J].Inorg.Chem., 2011, 50(8): 3816.
[7] Angamuthu R, Byers P, Lutz M,etal. Electrocatalytic CO2conversion to oxalate by a copper complex [J].Science, 2010, 327(5963): 313.
[8] Tsutsumi Y, Yamakawa K, Yoshida M,etal. Bifunctional organocatalyst for activation of carbon dioxide and epoxide to produce cyclic carbonate: betaine as a new catalytic motif [J].Org.Lett., 2010, 12(24): 5728.
[9] Bhavani A G, Kim W Y, Lee J S. Barium substituted lanthanum manganite perovskite for CO2reforming of methane [J].ACSCatal., 2013, 3(7): 1537.
[10] Sarusi I, Fodor K, Baan K,etal. CO2reforming of CH4on doped Rh/Al2O3catalysts [J].Catal.Today, 2011, 171(1): 132.
[11] Chen H L, Chen H T, Ho J J. Density functional studies of the adsorption dissociation of CO2molecule on Fe(111) surface [J].Langmuir, 2010, 26(2): 755.
[12] Liu C, Cundari T R, Wilson A K. CO2reduction on transition metal (Fe, Co, Ni, and Cu) surfaces: In comparison with homogeneous catalysis [J].J.Phys.Chem. C, 2012, 116(9): 5681.
[13] Poully B, Bergeat A, Hannachi Y. Theoretical study of the mechanism and rate constant of the B+CO2reaction [J].J.Phys.Chem. A, 2008, 112(35): 8148.
[14] Wu S Y, Ho J J. Adsorption, dissociation, and hydrogenation of CO2on WC(0001) and WC-Co alloy surfaces investigated with theoretical calculations [J].J.Phys.Chem. C, 2012, 116(24): 13202.
[15] Cheng Z, Sherman B J, Lo C S. Carbon dioxide activation and dissociation on ceria (110): A density functional theory study [J].J.Chem.Phys., 2013, 138(1): 014702.
[16] Tan S J, Zhao Y, Zhao J,etal. CO2dissociation activated through electron attachment on reduced rutile TiO2(110)-1×1 surface [J].Phys.Rev. B:Condens.Matter, 2011, 84(15): 155418.
[17] Lee J, Sorescu D C, Deng X Y. Electron-induced dissociation of CO2on TiO2(110) [J].J.Am.Chem.Soc. , 2011, 133(26): 10066.
[18] Novoselov K S, Jiang Z, Zhang Y,etal. Room-temperature quantum Hall effect in graphene [J].Science, 2007, 315(5817): 1379.
[19] Castro Neto A H, Guinea F, Peres N M R,etal. The electronic properties of graphene [J].Rev.Mod.Phys., 2009, 81(1): 109.
[20] Chi M, Zhao Y P. Adsorption of formaldehyde molecule on the intrinsic and Al-doped graphene: A first principle study [J].Comput.Mater.Sci., 2009, 46(4): 1085.
[21] Lv Y A, Zhuang G L, Wang J G,etal. Enhanced role of Al or Ga-doped graphene on the adsorption and dissociation of N2O under electric field [J].Phys.Chem.Chem.Phys., 2011, 13(27): 12472.
[22] Zhao J X, Chen Y, Fu H G. Si-embedded graphene: an efficient and metal-free catalyst for CO oxidation by N2O or O2[J].Theor.Chem.Acc., 2012, 131: 1242.
[23] Yu S S, Zheng W T, Wang C,etal. Nitrogen/boron doping position dependence of the electronic properties of a triangular graphene [J].ACSNano, 2010, 4(12): 7619.
[24] Frisch M J, Trucks G W, Schlegel H B,etal. Gaussian 03, Revision A. 1. Pittsburgh, PA: Gaussian, Inc., 2003.
[25] Han D M, Dai G L, Chen H,etal. DFT study of the reactions of Mo and Mo+ with CO2in gas phase [J].J.Chem.Sci., 2011, 123(3): 299.
[26] Han D M, Dai G L, Chen H,etal. Theoretical study on the reactions of Nb and Nb+with CO2in gas phase [J].Int.J.QuantumChem., 2011, 111(12): 2898.
[27] Fergusson A I.IdentifyingCO2dissociationpathwaysonsteppedandkinkedcoppersurfacesusingfirstprinciplescalculations[D]. Atlanta: Georgia Institute of Technology, 2012.
Theoretical study of the dissociation of CO2catalyzed by Cr/Ti-doped graphene
SHI Long-Xian, YANG Zhuan-Xia, FAN Mei-Ling, WANG Luo-Xin
(College of Materials Science & Engineering, Wuhan Textile University, Wuhan 430200, China)
In order to screen the highly efficient catalyst for the reduction of CO2molecule, the B3LYP method in combination with the 6-31G(d) basis set was used to calculate the adsorption of CO2molecule on surfaces of the Cr-doped graphene and Ti-doped graphene. Meanwhile, the CO2dissociation mechanism catalyzed by the Cr-doped graphene and Ti-doped graphene was also considered with the same calculation method. Results show that the process is exothermal for the CO2adsorption on the doped-graphene surface. The adsorption energies are 28.3 kcal/mol and 7.7 kcal/mol for the CO2adsorption on the Cr-doped graphene and Ti-doped graphene surface, respectively. The dissociation of CO2catalyzed by the Cr/Ti-doped graphene follows the insertion-elimination mechanism. The calculated activation energies of the C-O bond dissociation are 15.5 kcal/mol and 7.8 kcal/mol for the Cr and Ti-doped graphene, respectively. Our calculation results would be a guideline for the design of catalyst for the CO2reduction.
Doped-graphene; CO2; Dissociation; DFT
施龙献(1992—),男,浙江丽水人,主要从事功能高分子材料和理论化学研究. E-mail: wsxianjiayou@163.com
王罗新. E-mail: wanglx@wtu.edu.cn
103969/j.issn.1000-0364.2015.10.008
O643
A
1000-0364(2015)05-0763-06
投稿日期:2014-03-01
