苯的硝基和叠氮基衍生物热力学性质的构效关系
2015-03-23刘晓静何伟平
刘晓静, 何伟平, 黄 菊
(1.徐州工业职业技术学院化学工程技术学院, 徐州 221140; 2.徐州工程学院化学化工学院, 徐州 221111)
苯的硝基和叠氮基衍生物热力学性质的构效关系
刘晓静1, 何伟平1, 黄 菊2
(1.徐州工业职业技术学院化学工程技术学院, 徐州 221140; 2.徐州工程学院化学化工学院, 徐州 221111)
苯的硝基和叠氮基衍生物是一类重要的含能材料,为了揭示其热力学性质与分子结构之间的关系,采用第一性原理进行了计算研究.通过计算平衡电负性连接指数,结合分子结构描述符,对苯的硝基和叠氮基衍生物的热力学性质建立了构效关系模型.模型检验结果表明,构建的模型具有良好的稳健性和预测能力,所得模型为苯的硝基和叠氮基衍生物的爆轰参数计算和分解机理研究提供了一种快速的热力学性质预测方法.
构效关系; 热力学性质; 计算机模拟; 苯; 衍生物; 预测
1 引 言
由于分子结构中含有丰富的N-N、C-N等高能化学键,以苯环为母体的硝基和叠氮基衍生物已成为含能材料的研究热点之一[1,2].相关爆轰性能的研究工作有:1,3,5-三氨基-2,4,6-三硝基苯(TATB)生成焓的计算[3], 混合体系三硝基甲苯(TNT)+黑索今(RDX)的爆轰性质估算[4],适用于TATB、RDX和奥克托今(HMX)全原子力场的建立和验证[5],苯的硝基和叠氮基衍生物爆轰性能和稳定性能的预测[6]等.同时研究者对其分解反应机理也进行了大量研究,相关工作包括:TNT、2,4-二硝基甲苯高温热分解过程的研究[7-9],硝酸对硝基苯自加速分解影响的研究[10],TATB粉末在冲击波下分解机理的研究等[11].
如前所述,目前对苯的硝基和叠氮基衍生物虽已进行了一定的研究,但实验测定各种性能非常复杂,尤其热力学性质的系统化研究极为少见.鉴于定量结构-性质相关性(QSPR)现已广泛应用于化学领域[12,13],且通过第一性原理可以准确预测分子的物理化学性质[14-16].本研究运用第一性原理,通过建立构效关系模型,揭示了热力学性质与分子结构的关系, 所得模型可方便地预测热力学性质,进而得出苯的硝基和叠氮基衍生物热力学性质的共性规律,为爆轰性能和分解机理的研究提供依据.
2 热力学数据与分子描述符
2.1 热力学数据

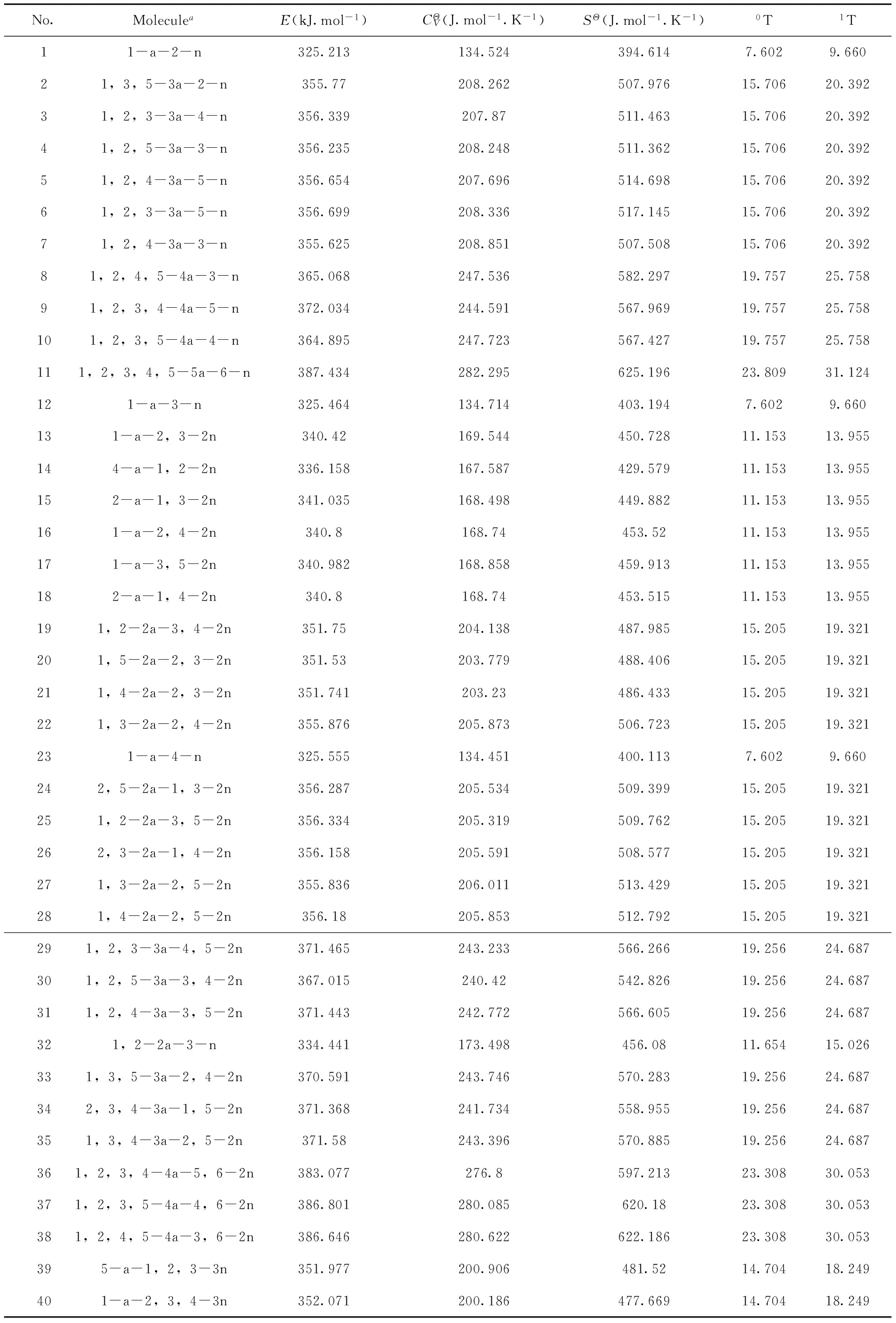
表1 苯的硝基和叠氮基衍生物的热力学性质
注:a—叠氮基,n—硝基;2a—两个叠氮基,2n—两个硝基;依次类推
2.2 分子描述符
考虑到分子的热力学性质与成键原子的种类和连接顺序密切相关,根据平衡电负性原理[20],本研究定义原子特征值如式(1)所示.
ti=(χiA·Ji+∑χG)/(1+∑l)/χC
(1)
式中,χiA表示i原子的Pauling电负性,Ji为修正系数(氮原子JN=1.53,氧原子JO=1),∑χG表示与i原子直接相连基团的电负性之和,∑l表示与i原子直接相连的基团数之和,χC为碳原子的Pauling电负性.其中对∑χG定义见式(2):
(2)
借鉴电负性连接指数形式[21],定义0阶、1阶平衡电负性连接指数分别为式(3)式(4)所示:
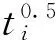
(3)
1T=∑(titi-1)0.5
(4)
其中,i-1表示与i直接相连的原子.为了简化计算,本研究仅计算硝基和叠氮基部分的0T和1T.


图1 1-叠氮基-2-硝基苯的分子结构Fig.1 Molecular structure of 1-azido-2-nitro-benzene
本研究采用Visual Basic 6.0软件自编程序对所有91个分子结构进行计算,并将各分子的0T、1T值列于表1.此外由于分子的硝基数Nn和相邻硝基对数Bn对热力学性质也会造成一定的影响,故将其与0T和1T均作为分子描述符进行构效关系研究.
3 模型的建立与分析
3.1 自变量的选择
各自变量不应有明显的相关性,否则应将多余的自变量删除,以便重新拟合模型.为了评价模型中各自变量的多重相关性,引入方差膨胀因子VIF,见式(5):
VIF=1/(1-R2)
(5)
式中,R为某一变量与余下变量的相关系数.若VIF<5,则表明变量间没有明显的自相关性;若5
由表2可见,0T、1T和Nn完全线性相关,剔除0T或1T均可实现剩余变量VIF<5.但是,剔除Nn则需要进一步剔除变量,从而会导致判定系数(调整判定系数)下降,故不宜采用.本研究中不妨剔除0T,确定1T、Nn和Bn作为自变量进行热力学性质建模.
3.2 模型建立过程

表2 分子描述符的膨胀因子
同时,本研究引入Akaike信息判据(AIC)和Kubinyi函数(FIT),以确定最终的QSPR模型,其计算公式如式(6)式(7)所示:
AIC=RSS×(n+b)/(n-b)2
(6)
FIT=R2(n-b-1)/[(n+R2)(1-R2)]
(7)
式中,RSS为方差和,n为化合物数,b为变量数.要求仅当所增加的变量能够减少AIC值或提高FIT值时才在原模型中增加该变量.AIC值越小,FIT值越大,则所建模型越稳定,预测能力越高.利用式(6)、式(7)计算出AIC、FIT值列于表3.
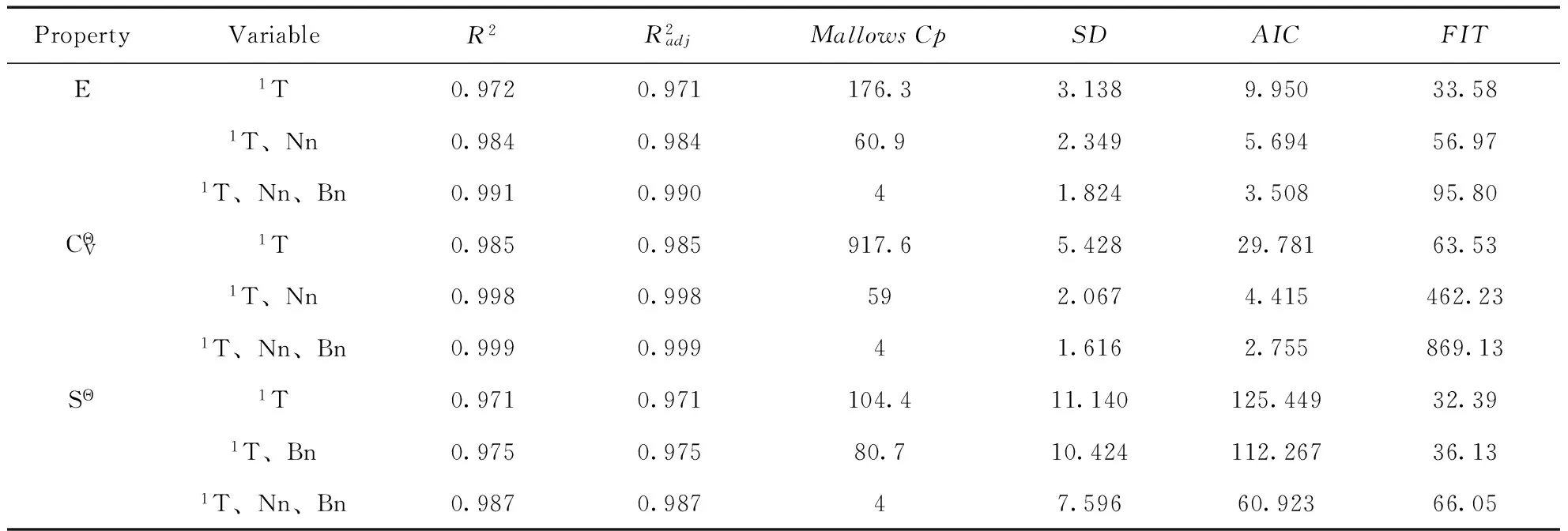
表3 最佳变量子集回归结果
根据回归结果和AIC值、FIT值判定原则,均可确定各热力学性质模型的自变量取{1T、Nn、Bn}组合为最佳.为了使所得预测模型具有较高的可信度,一般遵循如下经验规则:n/b≥5,本研究中n=91,b=3,因此变量数满足要求.
3.3 模型分析
构效关系回归模型最终结果见式(8)~式(10).
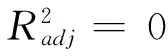
(8)
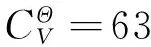
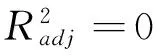
(9)
SΘ=291+10.41T-12.3Bn+9.36Nn
(10)
得到的3个构效关系模型,其调整判定系数均在0.98以上,属于优级相关.将估算值和理论值进行关联,二者大多比较吻合,见图2~图4.
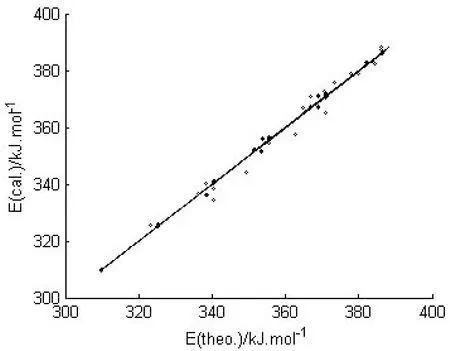
图2 E的估算值和理论值的相关性Fig.2 Relationship between calculated and theoretical E
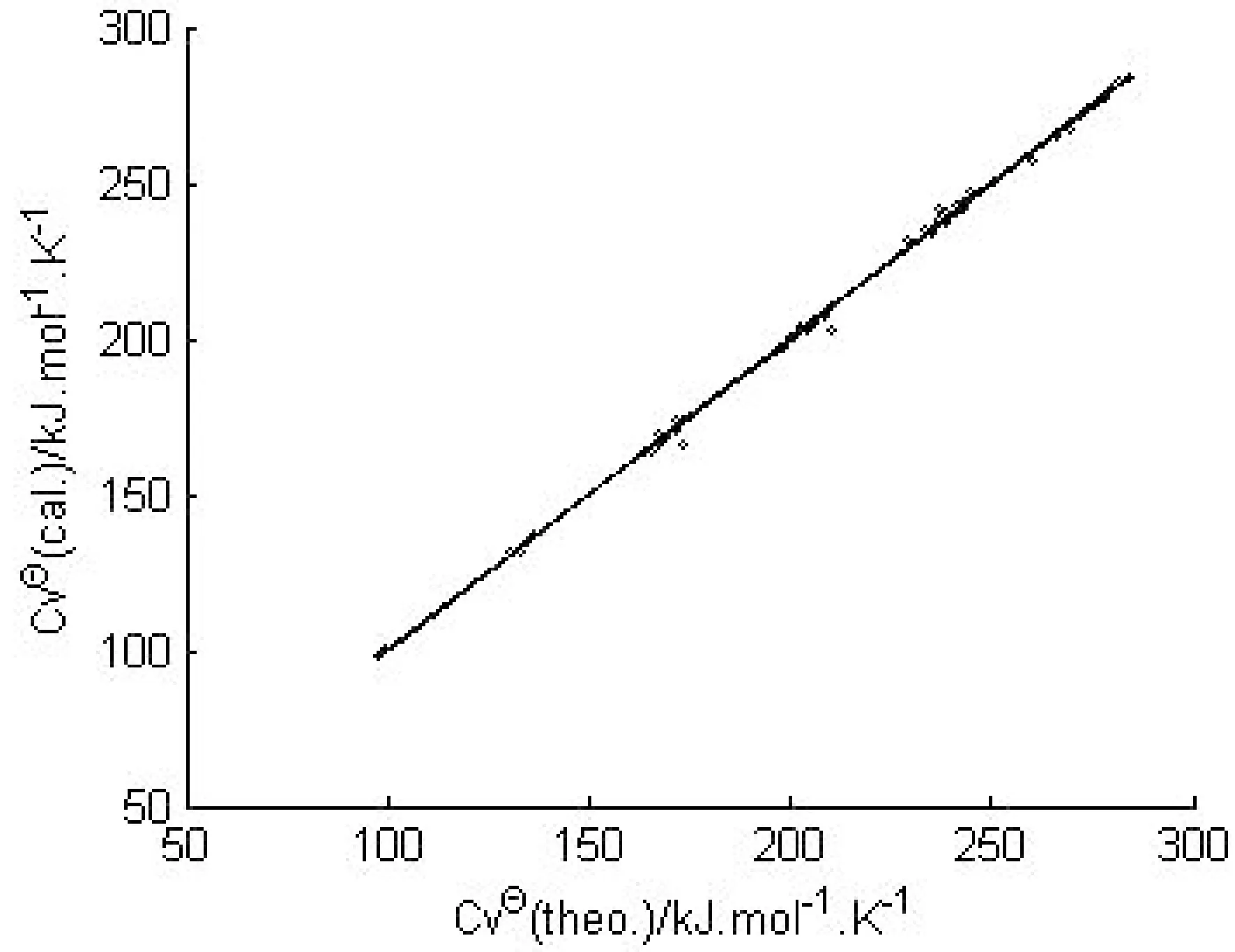
图的估算值和理论值的相关性Fig.
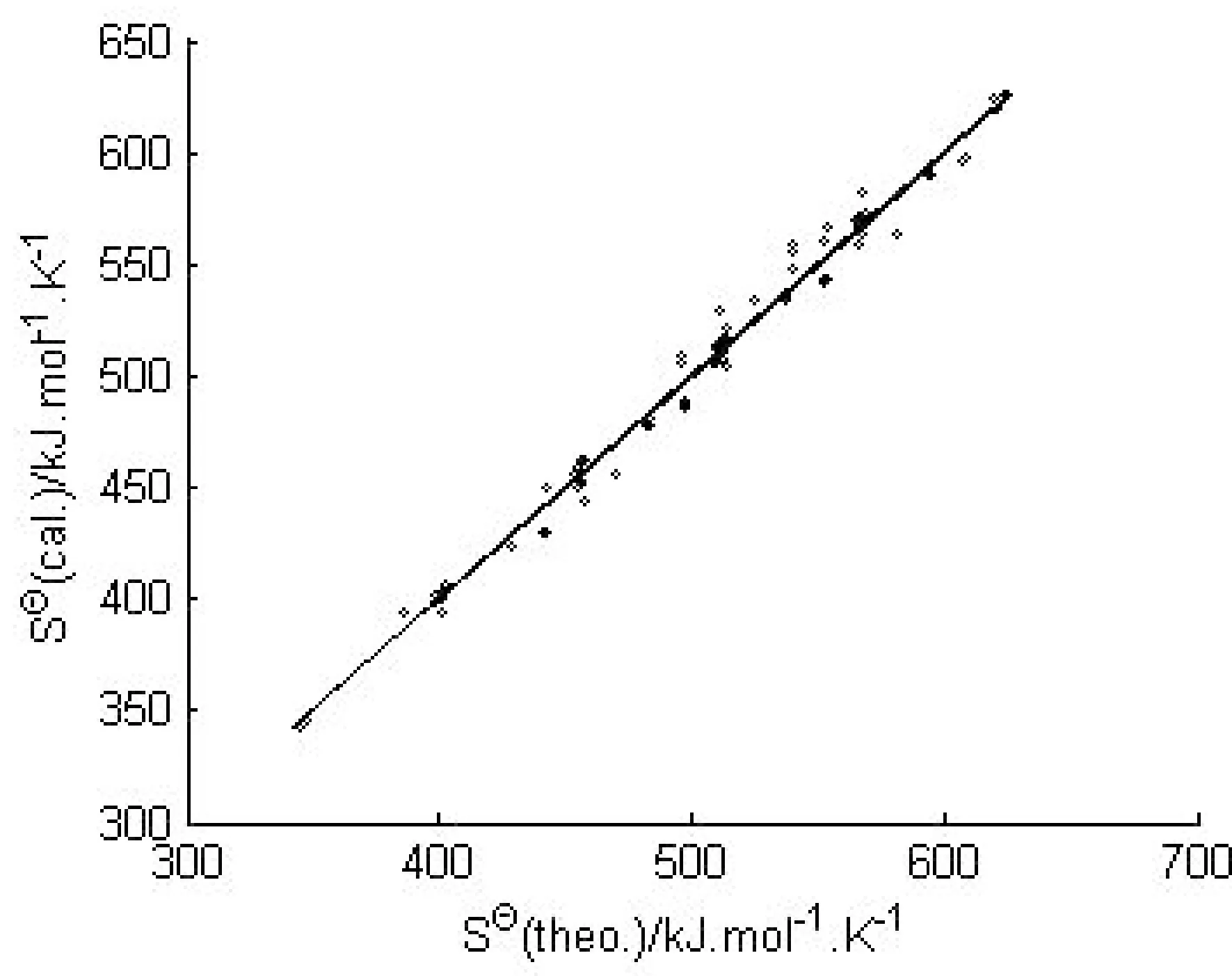
图4 SΘ的估算值和理论值的相关性Fig.4 Relationship between calculated and theoretical SΘ
4 模型检验
4.1 稳健性检验
为了检验模型的稳健性,以式(8)~式(10)中调整判定系数最低的SΘ为例说明.从表1中的91个分子中,依次抽出序号为1、7、13、…、85、91共16个分子作预测集(同样方法剔除2、8、14、…、86、91,依次类推),用余下的75个分子作训练集进行建模.对训练集进行最佳变量选择,以确定最佳变量子集.
4.2 预测能力
利用每个模型对相应被保留的16个分子的SΘ进行预测,得到的预测值与理论值较为吻合,说明提出的分子描述符用于预测苯的硝基和叠氮基衍生物的热力学性质是合理的.限于篇幅,这里以第1组为例,使用对应的模型对保留的16个分子进行预测(见表6).
由表6可见,预测误差最大的为1,2,3,4,5-五硝基苯(-3.93%),其预测值低于理论值,结合式(10)可以看出,这可能是由于其分子结构含有较多的相邻硝基引起的.此外,所有预测结果与理论值的相对误差均低于5%,说明本研究构建的模型预测能力较强.
5 结 论
本文根据平衡电负性指数0T和1T、硝基数Nn、相邻硝基对数Bn等分子描述符,经过变量筛选,最终确定1T、Nn和Bn作为模型输入参数,构建苯的硝基和叠氮基衍生物的热力学构效关系模型,获得了满意的调整判定系数,所建模型具有良好的稳定性和预测能力.

表4 SΘ与分子描述符的最佳变量子集回归结果
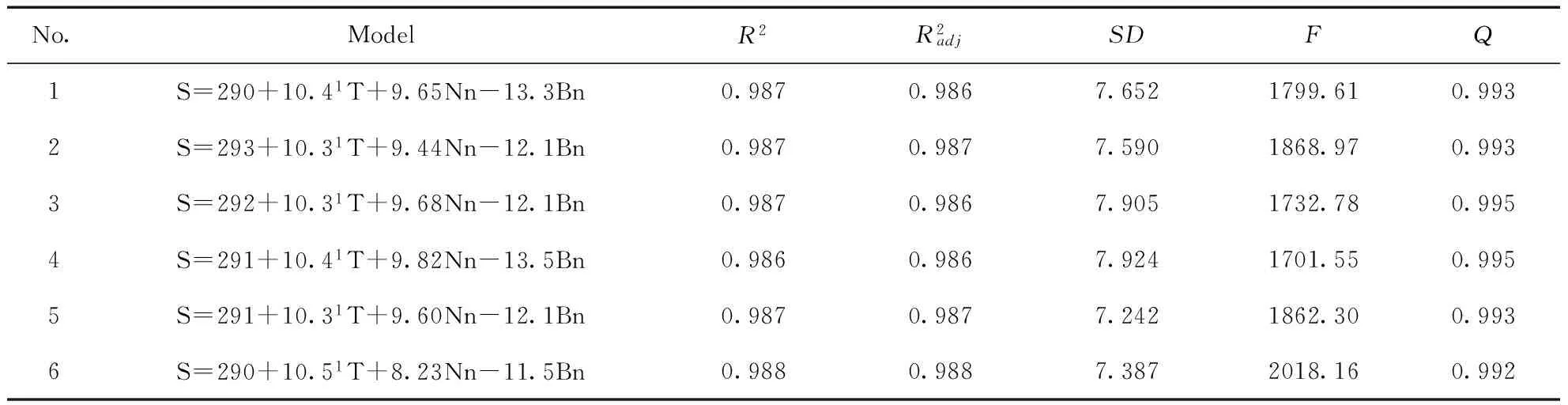
表5 训练集SΘ的回归结果
表6 苯的硝基和叠氮基衍生物的SΘ预测
Table 6 Predicting results forSΘof nitro and azido derivatives of benzene

No.MoleculeTheo.Pre.Error(%)11-a-2-n394.614400.2891.4471,2,4-3a-3-n507.508512.1810.92131-a-2,3-2n450.728441.447-2.06191,2-2a-3,4-2n487.985497.3931.93251,2-2a-3,5-2n509.762510.6580.18311,2,4-3a-3,5-2n566.605566.6040.00371,2,3,5-4a-4,6-2n620.18622.5500.38431,2-2a-4-n460.799456.235-0.99492,3-2a-1,4,5-3n559.986551.816-1.46551,2,4-3a-3,5,6-3n596.976607.7621.81611,4-2a-2,3,5,6-4n590.311592.9740.45671,2-2a403.645401.812-0.45731,3,5-3a463.182457.758-1.17791,2,3,5-4n479.862481.0830.25851,2,3,4,5-5n529.802508.975-3.93911,5-2a-2,4-2n508.267510.6580.47
(1)定义和构建了平衡电负性连接指数,模型结果表明,该指数能较好地预测相关的热力学性质.通过增加硝基数和相邻硝基对数这两个分子描述符,提高了模型的稳定性和预测能力.
[1] Kakar S,etal. Electronic of the energetic material 1, 3, 5- triamino -2, 4, 6- trinitrobenzen[J].Phys.Rev., 2000, B62(23): 1566.
[2] Liu X F, Xu W G, Lu S X. DFT theoretical study on nitrogen-rich compounds C6H6-n(N3)n(n=1-6)[J].ChemicalJournalofChineseUniversities, 2009, 30(7): 1406 (in Chinese)[刘晓芳, 徐文国, 卢士香. 叠氮化合物C6H6-n(N3)n(n=1~6)的密度泛函理论研究[J].高等学校化学学报, 2009, 30(7): 1406]
[3] Huang Z, Chen B, Liu F S. Quantum calculation for the enthalpy of formation of TATB[J].JournalofAtomicandMolecularPhysics, 2004, 21(3): 499 (in Chinese)[黄整, 陈波, 刘福生. TATB生成焓的量子力学计算[J]. 原子与分子物理学报, 2004, 21(3): 499]
[4] Niu X Q, Zhang J G, Feng X J,etal. Theoretical investigation on intermolecular interactions between the ingredients TNT and RDX of composition B[J].ActaChim.Sinica, 2011, 69: 1627 (in Chinese)[牛晓庆, 张建国, 冯晓军, 等. B炸药主要组分TNT和RDX分子间相互作用的理论研究[J]. 化学学报, 2011, 69: 1627]
[5] Jin Z, Liu J, Wang L L,etal. Development and validation of an all-atom force field for the energetic materials TATB, RDX and HMX[J].ActaPhys. -Chim.Sin., 2014, 30 (4): 654 (in Chinese)[金钊, 刘建, 王丽莉, 等. 适用于TATB, RDX, HMX含能材料的全原子力场的建立与验证[J].物理化学学报, 2014, 30 (4): 654]
[6] Du H C, Xu X J, Liu Y,etal. Theoretical studies on the nitro and azido derivatives of benzene[J].ActaChimicaSinica, 2011, 69(3): 269 (in Chinese)[杜洪臣, 许晓娟, 刘彦, 等. 苯的硝基和叠氮基衍生物的理论研究[J]. 化学学报. 2011, 69(3): 269]
[7] Liu H, Dong X, He Y H. Reactive molecular dynamics simulations of carbon-containing clusters formation during pyrolysis of TNT[J].ActaPhys. -Chim.Sin., 2014, 30 (2): 232 (in Chinese)[刘海, 董晓, 何远航. TNT高温热解及含碳团簇形成的反应分子动力学模拟[J].物理化学学报, 2014, 30 (2): 232]
[8] Yang T, Chen L P, Chen W H,etal. Experimental method on rapid identification of autocatalysis in decomposition reactions[J].ActaPhys. -Chim.Sin., 2014, 30 (7): 1215 (in Chinese)[杨庭, 陈利平, 陈网桦, 等. 分解反应自催化性质快速鉴别的实验方法[J]. 物理化学学报, 2014, 30 (7): 1215]
[9] Bao S L, Chen W H, Chen L P,etal. Identification and thermokinetics of autocatalytic exothermic decomposition of 2, 4-dinitrotoluene[J].ActaPhys. -Chim.Sin., 2013, 29 (03): 479 (in Chinese)[鲍士龙, 陈网桦, 陈利平, 等. 2, 4-二硝基甲苯热解自催化特性鉴别及其热解动力学[J]. 物理化学学报, 2013, 29 (03): 479]
[10] Ruan J F, Sun J H, Guo S,etal. Influence mechanism of nitric acid on the thermal stability of nitrobenzene[J].CIESCJournal, 2013, 5: 1526 (in Chinese)[阮继锋, 孙金华, 郭耸, 等. 硝酸对硝基苯热稳定性的影响机理[J]. 化工学报, 2013, 5: 1526]
[11] Carter Jeffrey A, Zaug Joseph M, Nelson A J,etal. Ultrafast shock compression and shock-induced decomposition of 1,3,5-triamino-2,4,6-trinitrobenzene subjected to a subnanosecond-duration shock: An analysis of decomposition products[J].TheJournalofPhysicalChemistryA, 2012, 116(20): 4851.
[12] Jin H,Wang X H,Yang F,etal. QSRR/QSPR models of physicochemical properties of polybrominated diphenyl ethers[J].CIESCJournal, 2014, 65(3): 797(in Chinese)[金浩, 王星皓, 杨芬, 等. 多溴联苯醚理化性质的定量构效关系[J]. 化工学报, 2014, 65(3): 797]
[13] Du X H. Physicochemical property of polybrominated diphenyl ethers by new path location index and neural network[J].CIESCJournal, 2014, 65(4): 1169(in Chinese)[堵锡华. 用新的路径定位指数和神经网络研究多溴联苯醚理化性质[J]. 化工学报, 2014, 65(4): 1169]
[14] Bode B M, Gordon M S. MacMolPlt: A graphical user interface for GAMESS[J].J.Mol.GraphicsMod., 1998, 16: 133.
[15] Li X M, Zhang J P, Mao Y. Charge transport and thermodynamic properties of triphenylence discotic liquid crystals with acetylamino chain[J].JournalofAtomicandMolecularPhysics, 2014, 31(6): 868 (in Chinese)[李雪梅, 张建平, 毛焱. 含乙酰胺基链苯并菲盘状液晶分子的电荷传输性质与热力学性质[J]. 原子与分子物理学报, 2014, 31(6): 868]
[16] Dong G X, Ge S H, Li D H. Density functional theory study on the temperature of nitromethane molecule under pressure[J].JournalofAtomicandMolecularPhysics, 2015, 32(2): 181(in Chinese)[董光兴, 葛素红, 李德华. 高压下硝基甲烷分子温度的密度泛函计算[J]. 原子与分子物理学报, 2015, 32(2): 181]
[17] Li B, Ren W. Thermodynamic properties of LiH and the effect of internal motion of LiH molecules on the thermodynamic properties of system[J].JournalofAtomicandMolecularPhysics, 2014, 31(5): 795(in Chinese)[黎波, 任维义. LiH的热力学性质及分子内部运动对体系热力学性质的影响[J]. 原子与分子物理学报, 2014, 31(5): 795]
[18] Granovsky Alex A, Firefly version 8.0, www ttp://classic.chem.msu.su/gran/firefly/index.html.
[19] Schmidt M W, Baldridge K K, Boatz J A,etal. General atomic and molecular electronic structure system[J].J.Comput.Chem. 1993, 14: 1347.
[20] Nie C M, Dai Y M, Wen S N,etal. Topological homologous regularity for additive property of Alkanes[J].ActaChim.Sinica, 2005, 63(15): 449(in Chinese)[聂长明, 戴益民, 文松年, 等. 烷烃加和型性质的拓扑同系递变规律研究[J]. 化学学报, 2005, 63(15): 449]
[21] Du X H. QSPR study on thermodynamic properties of polybrominated dibenzofurans and polybrominated dibenzothiophenes[J].CIESCJournal, 2010, 61(12): 3059(in Chinese)[堵锡华. 多溴代二苯并呋喃/噻吩热力学性质的定量构效关系[J]. 化工学报, 2010, 61(12): 3059]
QSPR study on thermodynamic properties of the nitro and azido derivatives of benzene
LIU Xiao-Jing1, HE Wei-Ping1, HUANG Ju2
(1.School of Chemical Engineering, Xuzhou College of Industrial Technology, Xuzhou 221140, China;2.School of Chemistry & Chemical Engineering, Xuzhou Institute of Technology, Xuzhou 221111, China)
The nitro and azido derivatives of benzene is a kind of important energy material, to reveal the relationships between thermodynamic properties and molecular structures of them, first-principles theory was adopted to calculate the thermodynamic properties. Based on the topological indexes of balanced electronegative connection and the molecular structure descriptors, with multiple linear regressions, the quantitative structure-property relationships between molecular structures and the thermodynamic properties were constructed. The leave-one out cross validation method was adopted to verify the stability and the prediction ability of each model, and the result shows quite satisfactory. It has been demonstrated that the models have satisfactory stability and good predictability. The models provide a rapid method for predicting thermodynamic properties to calculate the detonation parameters or to reveal the decomposition mechanism.
Quantitative structure-property relationship; Thermodynamic properties; Computer simulation; Benzene; Derivative; Prediction
刘晓静(1981—),女,内蒙古赤峰市人,硕士,讲师,主要研究含能材料性能等.E-mail: liuxj1981@163.com
103969/j.issn.1000-0364.2015.10.007
TJ55; TK421
A
1000-0364(2015)05-0754-09
投稿日期:2015-01-22
