CuCl催化下苯炔与环己烯乙炔偶联反应机理的理论研究
2015-03-23王晓岚周红平李来才
王晓岚, 周红平, 郑 妍, 李来才
(1.四川师范大学化学与材料学院, 成都 610066; 2.西南交通大学材料科学与工程学院, 成都 610031)
CuCl催化下苯炔与环己烯乙炔偶联反应机理的理论研究
王晓岚1, 周红平2, 郑 妍1, 李来才1
(1.四川师范大学化学与材料学院, 成都 610066; 2.西南交通大学材料科学与工程学院, 成都 610031)
利用密度泛函理论(DFT)研究了CuCl催化下苯炔与环己烯乙炔偶联反应的微观反应机理.在B3LYP/6-31+G* 基组水平上(Cu采用了赝势基组LanL2DZ)优化了反应过程中所有化合物的几何构型并计算了频率,通过能量、频率和振动方式确定了中间体和过渡态的真实性.此外,在同等基组水平上还运用了分子中的原子理论讨论了成键临界点的电荷密度的变化,运用了自然键轨道理论讨论了键的性质与轨道间的相互作用.为了提高计算精度, 在6-311+G* 基组水平上计算了反应机理中所有物质在气相及溶剂化下的单点能, 得到与 6-31+G* 基组计算相同的结论.结论表明CuCl对苯炔与1-乙炔基环己烯偶联反应起到了有效的催化作用,且计算所得结论与实验结果相符合.
密度泛函理论; 芳香化合物; CuCl催化; C-C偶联反应; 微观反应机理
1 引 言
芳香化合物的合成是近代有机合成中最重要研究领域,它被广泛的应用于生物制药、生物碱、农业用药等多个方面[1,2].现代工业中运用的染料和电子工业中的半导体材料很多也要用到芳香化合物.由于乙烯基能够发生多种化学转化, 如环氧化、双羟基化、臭氧开环以及卡宾加成等, 所以对含有乙烯基的炔对苯炔的不对称加成反应研究具有很高的应用价值[3].同时由于芳炔构建苯环结构或者多取代的芳烃的合成相当困难,所以由过渡金属催化芳炔变形为苯环结构或者多取代的芳烃作为一种有效的合成方法近年来受到了广泛关注[4-10].自从Pena成功用Pd催化合成出了三聚芳炔以来,迄今为止绝大多数的对于芳炔的变形研究都是采用了以Pd或者Ni及其相关化合物作为催化剂来完成[11].且近年来很多芳香化合物的合成都依然是运用Pd催化的Stille偶联反应和Suzuki偶联反应来完成,但其毒性大价格昂贵等缺点促使化学家们寻求更绿色、更经济的催化体系[12].因为芳炔其自身具有的亲电子特性,可以利用铜的乙炔化合物的亲核特性使其作为一种有效地结合芳炔的试剂,产生一种构建芳香族化合物的新的催化路径.且Cu盐催化的反应和Pd、Ni等过渡金属催化的C-C偶联反应相比较其更具有廉价、易得、稳定等特点,可以大规模应用在工业生产中[13, 14].更重要的是Cu金属性质比较温和而且配体简单,可以避免发生过渡金属催化反应中还原消除时发生的β-H消除反应,从而避免发生β-H底物的副反应,此外,还可以避免在反应中由过渡金属引起的双键移位现象[15, 16].因此,目前催化偶联领域中应用铜盐作为催化剂是一个非常热门的话题.如何更好地应用铜盐催化C-C键交叉偶联反应,不仅是过渡金属催化偶联反应领域中的一个新动向,也是绿色化学工业化进程中的一个具有挑战性的课题.通过与实验的比较,与以往采用Pd或者Ni及其相关化合物作为催化剂将芳炔和炔烃偶联起来的反应路径相比,我们发现Yoshida等在实验室里运用铜盐作为催化剂完成了与其以完全不同的合成方式来促使芳炔和炔烃偶联的反应[17, 18].本文通过研究苯炔与环己烯乙炔在CuCl催化下发生加成偶联反应的微观反应机理,分析最可能实现的反应路径,进一步了解此反应催化剂、配体作用的本质特征和反应的微观历程,为相关实验研究提供理论信息.
2 计算方法
利用密度泛函理论(DFT)的B3LYP方法,在6-31+G* 基组水平上(Cu采用了赝势基组LanL2DZ)对 CuCl催化下苯炔与1-乙炔基环己烯偶联反应过程中所有的反应物、中间体、过渡态和产物进行了结构优化,并在相同基组水平下对各构型进行了频率计算.运用自然键轨道(NBO)方法分析了中间体、过渡态的轨道间相互作用[19].反应公式如下图:
同时使用自洽反应场(SCRF)极化连续模型(IEF-PCM)模拟实验所使用的四氢呋喃(THF)溶剂效应, 采用6-311+G* 基组水平对反应机理中各化合物均进行了全参数优化,并使用相同基组计算了气相条件下的单点能.为了进一步的了解成键性质分析成键特征,采用AIM2000的程序包计算了对应的所有成键临界点(BCP)和成环临界点(RCP) 电荷密度,显示分子中原子的属性[20].所有计算均采用Gaussian03程序完成[21].
3 反应机理和能量分析
实验报道中, 在反应条件筛选时使用的溶剂为四氢呋喃THF, 因此在进行反应机理的理论研究时选用与实验一致的THF 作为溶剂.对在6-31+G* 基组下优化后的反应物、中间体、过渡态以及生成物的构型使用大基组6-311+G* 进行了单点能计算,气相和溶剂化条件下通过计算所得能量(E)、相对能量(Erel)及过渡态频率数值均已列于表1,能量(E)已包括零点能(ZPE)修正.
Scheme 1即为CuCl催化下的苯炔与1-乙炔基环己烯发生偶联反应机理图.将本文计算的CuCl催化下苯炔与1-乙炔基环己烯偶联反应的最可能的反应通道scheme1与文献[17]中通过实验所推断出的最可能的反应通道相比较,可以看出本文与yoshida的观点结论相一致.即1-乙炔基环己烯先与催化剂CuCl发生反应,结合第一个苯炔分子,然后通过亲核加成反应连接第二分子苯炔,生成最后的产物.整个偶联反应过程中所涉及的所有反应、中间体、过渡态及产物的构型见图1.
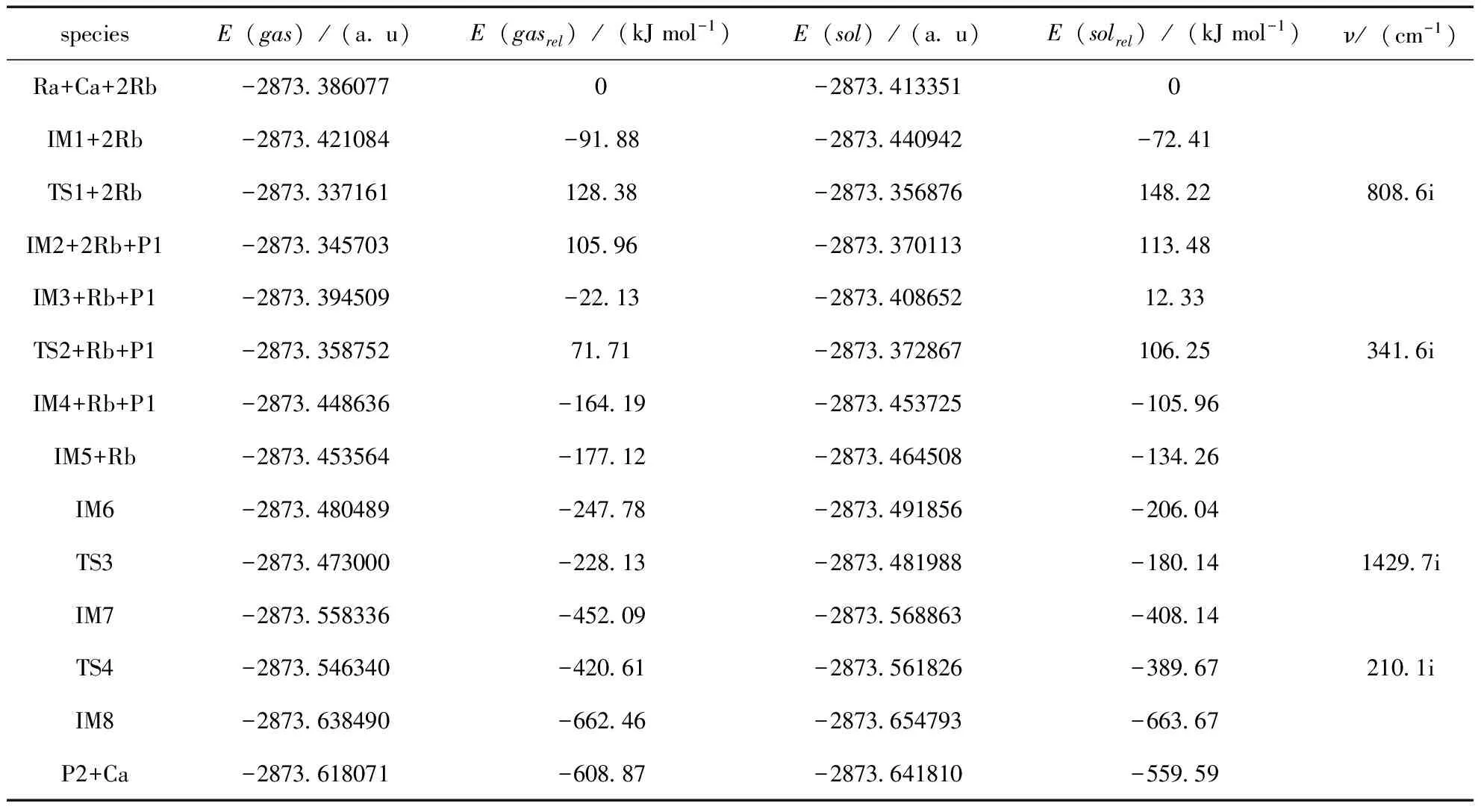
表1 反应各驻点能量E (a.u.)、相对能量Erel (kJ mol-1)及各过渡态频率v (cm-1)

Scheme 1 CuCl 催化下苯炔与1-乙炔基环己烯的偶联反应微观流程图
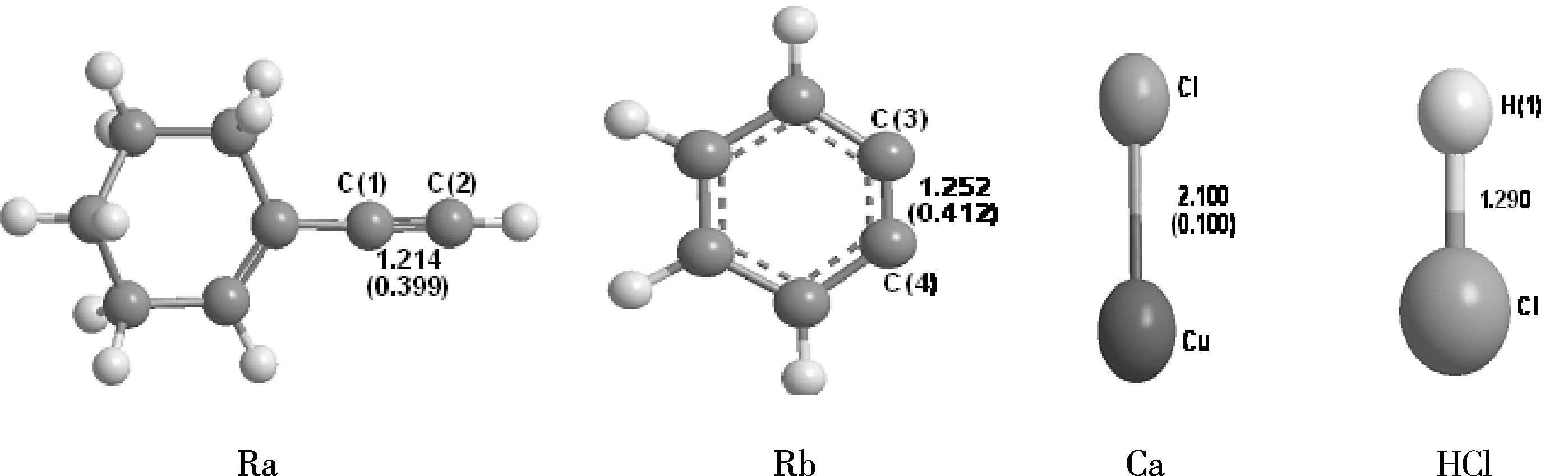
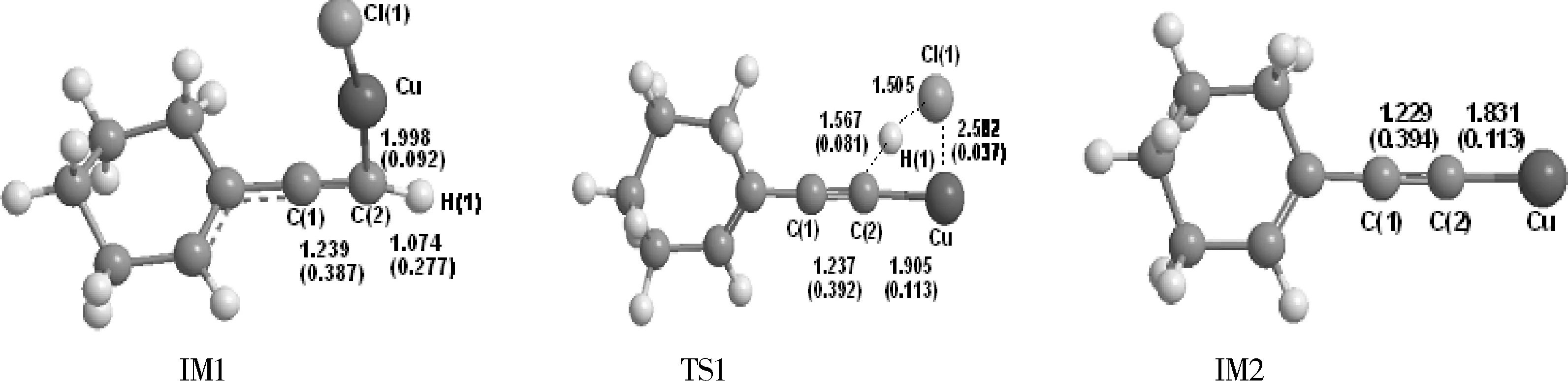

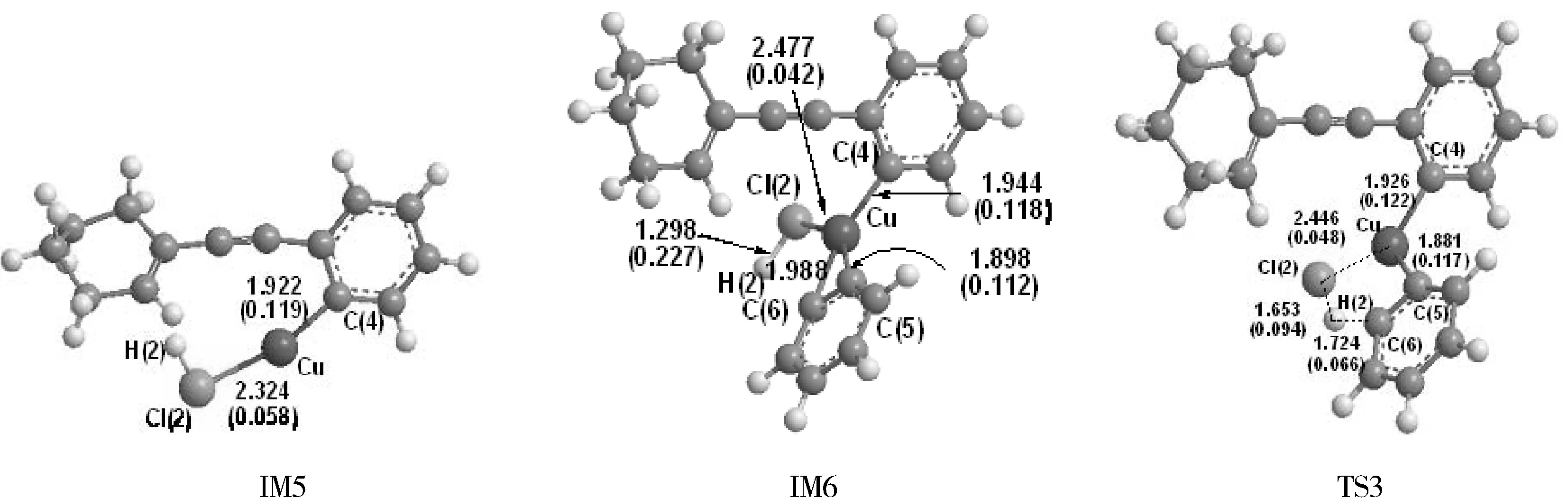
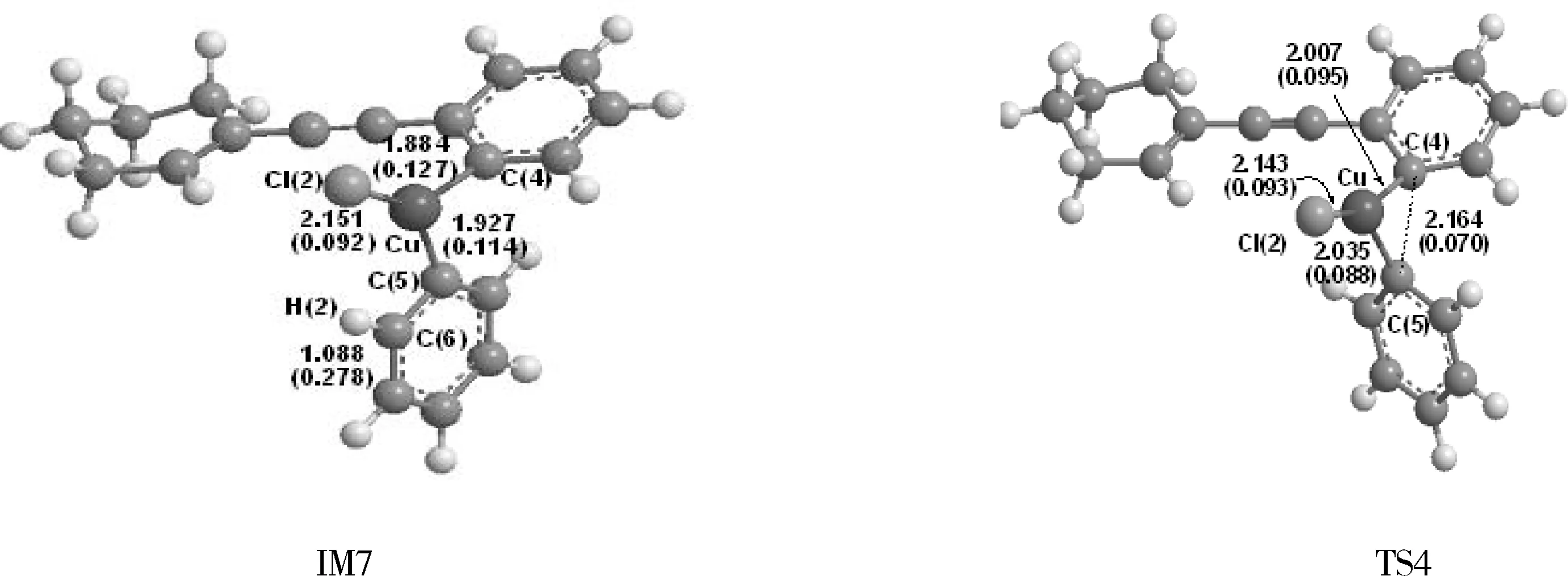

图1 反应过程化合物的几何优化构型Fig.1 Optimized structures of the compounds (Bond length in Å,BCP(in brackets)in a.u.)
反应物Ra(1-乙炔基环己烯)中末端的C原子与催化剂CuCl中的Cu原子成键,生成IM1,体系能量降低91.88 kJ/mol.说明IM1的构型稳定存在,催化剂的吸附过程较容易完成.在理论计算中,IM1经由过渡态TS1脱去HCl生成IM2.但在实际的反应存在KF碱性溶剂, 在碱性条件下, HCl通过酸碱中和而脱离.计算得到的过渡态TS1只是个可能的过渡态, 实际反应是通过酸碱中和直接得到中间体IM2.在中间体IM2中,C(1)-C(2) 键长为1.229 Å,电荷密度为0.394 a.u.;C(2)-Cu键长和电荷密度分别为1.831 Å和0.113 a.u..C(2)-H(1)键、Cu- Cl(1)键断开,H(1)-Cl(1)成键后从化合物上脱落.中间体IM2与反应物Rb反应,苯炔中的C(3)、C(4)原子连接在Cu原子上,生成芳基铜化合物IM3,体系能量降低128.09 kJ/mol.IM3中的C(2)-Cu键长和电荷密度分别为1.867 Å和0.394 a.u.,与IM2相比,键长增大了0.036 Å,电荷密度增大了0.015 a.u..化合物中新成键的C(3)-Cu和C(4)-Cu的键长分别为1.963 Å和1.966 Å,BCP电荷密度皆为0.099 a.u..Cu原子的孤对电子轨道上的电子大幅度跃迁到其激发态Rydberg轨道,二阶稳定化能达到97.14 kJ/mol,原子化学反应活性增大,与苯炔顺利连接.苯炔中的C(3)-C(4)的成键轨道提供给Cu的孤对电子,对应的二阶稳定化能为22.66 kJ/mol.
IM3经由过渡态TS2生成IM4,这是Cu原子转移促使C-C耦合发生的过程.IM3至TS2的反应活化能为93.84 kJ/mol,而由过渡态TS2完成C-C耦合到稳定状态IM4,体系能量降低了235.90 kJ/mol.在TS2中,C(2)-Cu键长和电荷密度分别为1.893 Å和0.116 a.u.,C(2)-C(3)键长和电荷密度分别为2.101 Å和0.073 a.u.,C(3)-Cu和C(4)-Cu的键长分别为2.004 Å和1.939 Å,BCP电荷密度分别为0.067 a.u.和0.101 a.u..与IM3相比较, TS2中的C(1)-C(2) 键长、电荷密度几乎没有发生改变,C(3)-Cu键长增大了0.041 Å,C(2)-Cu键长增大了0.026 Å,C(4)-Cu的键长减少了0.027 Å.而C(2)原子和C(3)原子的距离由之前IM3中的3.823 Å减少至TS2中的2.101 Å,这说明C(2)-C(3)已经微弱成键.IM4中的C(2)-C(3)键长和电荷密度分别为1.429 Å和0.287 a.u.;C(4)-Cu键长和电荷密度分别为1.903 Å和0.122 a.u..C(2)-C(3)键成键的同时,C(3)-Cu、C(2)-Cu键断裂.通过对IM4的NBO分析,C(2)-C(3)键主要是SP-SP杂化轨道形成的σ键,电子占据数为1.97443个,该键相对能量为-0.68967 a.u.,由金属Cu催化的C(2)-C(3)偶联过程完成.
由于IM4的亲核性,一分子的HCl吸附其上形成IM5,再与一分子苯炔相反应,生成中间体IM6.在IM4生成IM5的过程中,Cl(2)原子吸附于Cu原子之上,Cu-Cl(2)键长和电荷密度分别为2.324 Å和0.058 a.u.,C(4)-Cu键长和电荷密度分别为1.922 Å和0.119 a.u..分子构型没有发生太大变化,体系能量降低了12.93 kJ/mol,吸附过程较容易完成.在IM5生成IM6的过程中,苯炔中的C(5)、C(6)原子连接在Cu原子上,体系能量降低了70.66 kJ/mol.化合物中新成键的C(5)-Cu和C(6)-Cu的键长分别为1.898 Å和1.988 Å,BCP电荷密度皆为0.112 a.u..
IM6中的H原子逐渐靠近苯炔分子的C(6),经由过渡态TS3,C(6)-Cu键、Cl(2)-H(2)断裂,C(6)-H(2)成键,生成中间体IM7,这是一个H转移的过程.IM6至TS3的反应活化能为19.65 kJ/mol,反应能垒较低,与IM6相比,TS3中C(4)-Cu键长减小了0.018 Å,BCP电荷密度增大了0.004 a.u.;C(5) -Cu键长减小了0.017 Å,电荷密度增大了0.005 a.u.;Cu- Cl(2)键长减小了0.001 Å,电荷密度增大了0.006 a.u.;Cl(2)-H(2)键长增大了0.355 Å,电荷密度减小了0.133 a.u..C(6)原子与Cu原子之间的距离由IM6中的1.988 Å变为TS3中的2.129 Å,趋于断开;C(6)原子与H(2)原子之间的距离由IM6中的3.329 Å变为TS3中的1.724 Å,逐渐成键.通过对TS3的NBO分析,C(5)-Cu键的电子占据数为1.62190个,相对能量为-0.36905 a.u..TS3生成IM7的过程中,体系能量降低了223.36 kJ/mol.C(4)-Cu键长和电荷密度分别为1.884 Å和0.127 a.u.;C(5)-Cu键长和电荷密度分别为1.927 Å和0.114 a.u.;Cu- Cl(2)键长和电荷密度分别为2.151 Å和0.092 a.u.;C(6)- H(2)键长和电荷密度分别为1.088 Å和0.278 a.u..C(6)-Cu键、Cl(2)-H(2)键完全断开,C(6)- H(2)成键,H原子的转移过程完成.中间体IM7中的C(4)原子与C(5)原子逐渐拉近,经由过渡态TS4,生成稳定中间体IM8,最后脱去催化剂生成产物P2,完成整个催化过程.在IM7到TS4的过程中,反应活化能为31.48 kJ/mol,反应能垒依然较低.在TS4中,C(4)-Cu键长和电荷密度分别为2.007 Å和0.095 a.u.;C(5)-Cu键长和电荷密度分别为2.035 Å和0.088 a.u.;Cu- Cl(2)键长和电荷密度分别为2.151 Å和0.092 a.u.;C(4)- C(5)键长和电荷密度分别为2.164 Å和0.070 a.u..与IM7相比较,Cu-Cl(2)键长与电荷密度变化均不大;C(4)-Cu键与C(5)-Cu键的键长分别增大了0.123 Å和0.108 Å,电荷密度分别减少了0.032 a.u和0.026 a.u;C(4)-C(5)键长由IM7中的2.475 Å变为2.164 Å,减小了0.311 Å.说明体系中的两个苯环在不断地靠拢,C(4)-C(5)在逐渐成键,C(4)-Cu与C(5)-Cu键逐渐断开,Cu原子在远离的同时往下方的苯环方向拉扯.TS4生成稳定中间体IM8的过程中,体系能量降低了241.85 kJ/mol.IM8中的C(4)-C(5)键长和电荷密度分别为1.495 Å和0.262 a.u.;Cu- Cl(2)键长和电荷密度分别为2.131 Å和0.094 a.u..与TS4相比较,IM8中的C(4)-C(5)和Cu- Cl(2)键的键长分别减小了0.669 Å和0.012 Å,电荷密度分别增大了0.192 a.u和0.001 a.u,Cu- Cl(2)键变化不大,C(4)-C(5)的成键完成了又一个C-C的耦合过程.而C(4)-Cu与C(5)-Cu的距离在IM8中达到了3.082 Å和2.212 Å,在Cu原子与C(4)原子C(5)原子之间的键逐渐断裂之后,Cu原子被朝苯环下方拉扯并最终与C(6)原子成键,键长为2.131 Å,电荷密度为0.069 a.u..对IM8的NBO分析结果可以得知,C(4)-C(5)的电子占据数为1.97242个,该键相对能量为-0.67391 a.u..IM8接着脱去催化剂CuCl,得到产物P2,至此,整个偶联反应过程完成.
CuCl催化下苯炔与1-乙炔基环己烯偶联反应的反应过程可能为:Ra+Ca →IM1 →TS1 →IM2(-P1+Rb) →IM3 →TS2 →IM4(+P1)→ IM5(+Rb)→ IM6→ TS3→ IM7→ TS4→ IM8→ P2+Ca.整个反应的速控步骤为IM3 →TS2 →IM4,反应活化能为93.84kJ/mol.结合表1所列在相同基组水平上溶剂化条件下理论计算所得的能量数据,可知能量变化趋势和气相条件下是一致的.
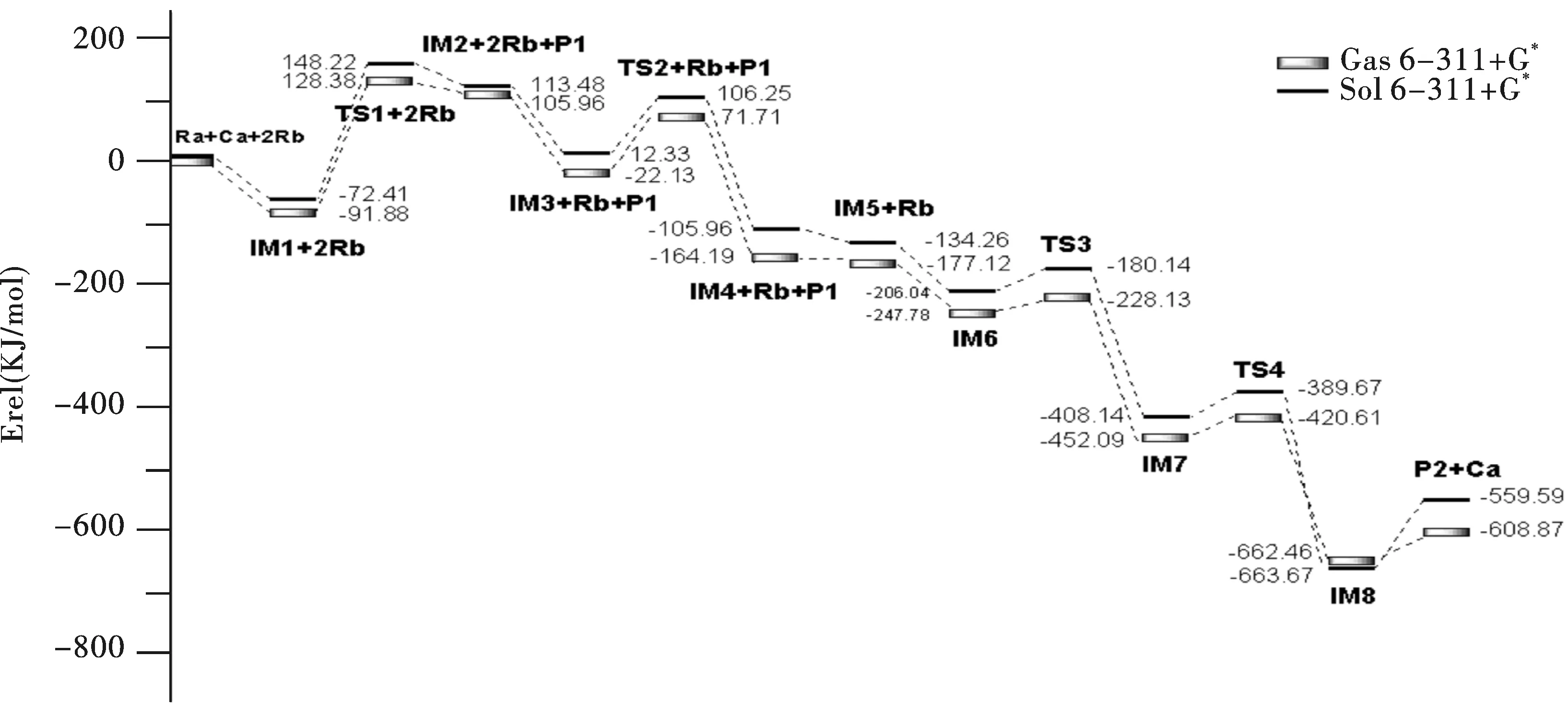
图 2 反应能级图Fig.2 Schematic of energy levels in the reaction
4 结 论
本文采用密度泛函理论(DFT)的B3LYP方法,在6-31+G* 基组水平上(Cu采用了赝势基组LanL2DZ)研究了CuCl催化下苯炔与1-乙炔基环己烯偶联反应的微观反应机理.同时使用自洽反应场(SCRF)极化连续模型(IEF-PCM)模拟实验所使用的四氢呋喃(THF)溶剂效应, 采用6-311+G* 基组水平对反应机理中各化合物均进行了全参数优化,并使用相同基组计算了气相条件下的单点能.结论如下:(1)在苯炔与1-乙炔基环己烯偶联反应中CuCl是一种有效的催化剂.(2)在芳炔与炔烃的偶联反应中,以铜盐做催化剂的反应机理与以Pd、Ni化合物做催化剂的反应机理完全不同.(3)在溶剂化条件下反应能量变化趋势和气相条件下是一致的.(4)计算所得该反应的微观机理与Yoshida等的实验结果、推断过程及实验结论相符合[17].
[1] Hassan J, Sévignon M, Gozzi C,etal. Aryl-aryl bond formation one century after the discovery of the Ullmann reaction[J].Chem.Rev., 2002, 102: 1359.
[2] Zhang G F, Zhao X B, Ding C R,etal. Process on direct catalytic oxidative coupling of arenes[J].Chin.J.Org.Chem., 2011, 31: 1736 (in Chinese) [张国富, 赵晓宝, 丁成荣, 等. 芳香化合物直接催化氧化偶联反应研究进展[J]. 有机化学, 2011, 31: 1736]
[3] Zhang D Y, Wang Q, Chen F X,etal. Asymmetric addition of 1-Ethynylcyclohexene to ketones catalyzed by a chiralC2-camphorsulfonamide ligand[J].Chem.J.Chin.Univ., 2008, 29: 1750 (in Chinese) [张东岩, 汪权, 陈福欣, 等. C2 轴对称樟脑磺酰胺基醇配体催化环己烯乙炔对酮的不对称加成反应[J]. 高等学校化学学报, 2008, 29: 1750]
[4] Tao L M, Liu W Q, Zhou Y. Study on the Sonogashira cross-couplings of aryl halides with terminal alkynes by Pd(OAc)2/TBAB[J].Chin.J.Syn.Chem., 2010, 18: 196 (in Chinese) [陶李明, 刘文奇, 周芸. Pd(OAc)2/TBAB催化卤代芳烃与末端炔烃的Sonogashira交叉偶联反应研究[J]. 合成化学, 2010, 18: 196]
[5] Huang J K, Nolan S P. Efficient cross-coupling of aryl chlorides with aryl grignard reagents (Kumada reaction) mediated by a palladium/imidazolium chloride system[J].J.Am.Chem.Soc., 1999, 121: 9889.
[6] Espinet P, Echavarren A M. The mechanisms of the Stille reaction[J].Angew.Chem.Int.Ed., 2004, 43: 4704.[7] Phapale V B, Cárdenas D J. Nickel-catalysed Negishi cross-coupling reactions: scope and mechanisms[J].Chem.Soc.Rev., 2009, 38: 1598.
[8] Daugulis O, Do H Q, Shabashov D. Palladium- and copper-catalyzed arylation of carbon-hydrogen bonds[J].Acc.Chem.Res., 2009, 42: 1074.
[9] Shu C L, Li B J, Shi Z J. Pd-catalyzed oxidative coupling with organometallic reagents via C-Hactivation[J].Chem.Commun., 2010, 46: 677.
[10] Yu J Q, Shi Z J. Topics in current chemistry[J]. 2010, 292: 165.
[11] Peńa D, Escudero S, Pérez D,etal. Efficient palladium-catalyzed cyclotrimerization of arynes: synthesis of triphenylene [J].Angew.Chem.Int.Ed., 1998, 37: 2659.
[12] Alonso F, Beletskaya I P, Yus M. Non-conventional methodologies for transition-metal catalysed carbon-carbon coupling: a critical overview. Part 2: The Suzuki reaction[J].Tetrahedron., 2008, 64: 3047.
[13] Wolter M, Nordmann G, Job G,etal. Copper-catalyzed coupling of aryl iodides with aliphatic alcohols[J].Org.Lett., 2002, 4(6): 973.
[14] Xu J H, Man Q S, Lin Y C,etal. Recent progress in copper catalyzed carbon-hetero cross-coupling reactions[J].Chin.J.Org.Chem., 2010, 30: 9(in Chinese) [徐建华, 蔄秋石, 林义成, 等. Cu催化碳杂偶联反应的新进展[J]. 有机化学, 2010, 30: 9]
[15] Kwong F Y, Buchwald S L. Mild and efficient copper-catalyzed amination of aryl bromides with primary alkylamines[J] .Org.Lett., 2003, 5(6): 793.
[16] Gelman D, Jiang L, Buchwald S L. Copper-catalyzed C-P bond construction via direct coupling of secondary phosphines and phosphites with aryl and vinyl halides[J].Org.Lett., 2003, 5(13): 2315.
[17] Yoshida H, Morishita T, Nakata H,etal. Copper-catalyzed 2:1 coupling reaction of arynes with alkynes[J].Org.Lett., 2009, 11(2): 373.
[18] Kang S K, Yoon S K, Kim Y M. Copper-catalyzed coupling reaction of terminal alkynes with aryl- and alkenyliodonium salts[J].Org.Lett., 2001, 3(17): 2697.
[19] Reed A E, Weinhold F, Curtiss L A,etal. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint[J].J.Chem.Rev., 1988, 88 (6): 899.
[20] Bader R W F.Atomsinmolecules:Aquantumtheory[M]. Oxford: Oxford University Press, 1990.
[21] Frisch M J, Trucks G W, Schlegel H B,etal. Gaussian 03, Revision A7. Pittsburgh PA: Gaussian Inc, 2003.
Theoretical investigation on the cross-coupling reaction mechanism of arynes with alkynes catalyzed by CuCl
WANG Xiao-Lan1, ZHOU Hong-Ping2, ZHENG Yan1, LI Lai-Cai1
(1.College of Chemistry and Material Science, Sichuan Normal University, Chengdu 610066, China;2.College of Materials Science and Engineering, Southwest JiaoTong University, Chengdu 610031, China)
The reaction mechanism of Arynes with Alkynes catalyzed by CuCl has been investigated by density functional theory (DFT). The geometries and the frequencies of reactants, intermediates, transition states, and products have been calculated at the B3LYP/6-31+G* level, and the LanL2DZ basis has been used as the extrabasis. The vibration analysis demonstrates that the authenticity of transition states, and the reaction processes are confirmed by the changes of charge density at bond-forming critical point which analyzed by the atoms in molecules theory. In addition, the nature bond orbital has been used to discuss the bond nature and orbital interactions at the same level. Meanwhile, the single point energy of the reaction process in gas and solvent at 6-311+G* level has been individually investigated with higher precision. The results indicate that the reaction mechanism and the change trend of correspondence energy at two different levels are consistent. The result of the theory study agrees with the experimental data, it indicates that the CuCl is an effective catalyst in this reaction.
Density functional theory; Aromatic compounds; CuCl-catalyze; C-C coupling reaction; Reaction mechanism
103969/j.issn.1000-0364.2015.02.006
2013-12-04
四川省教育厅重点基金项目(13ZA0150);四川师范大学重点资助项目
王晓岚(1984—),女,助教,硕士,主要从事应用量子化学研究. E-mail: 29757739@qq.com
李来才. E-mail: lilcmail@163.com
O641.3
A
1000-0364(2015)02-0207-07
