孤立条件下布洛芬分子手性转变过程的理论研究
2015-03-23邹晓威梅泽民王丽萍于天荣王佐成
邹晓威, 梅泽民, 王丽萍, 佟 华, 于天荣, 王佐成,4
(1.海口经济学院数理教研部, 海口 570100; 2.白城师范学院化学学院, 白城 137000;3.白城师范学院物理学院, 白城 137000; 4.吉林大学原子与分子物理研究所, 长春 130012)
孤立条件下布洛芬分子手性转变过程的理论研究
邹晓威1, 梅泽民2, 王丽萍3, 佟 华3, 于天荣3, 王佐成3,4
(1.海口经济学院数理教研部, 海口 570100; 2.白城师范学院化学学院, 白城 137000;3.白城师范学院物理学院, 白城 137000; 4.吉林大学原子与分子物理研究所, 长春 130012)
基于密度泛函理论的B3LYP方法,采用6-31+g(d,p)基组,对孤立条件下布洛芬分子的手性转变过程进行研究.通过寻找反应过程中包括过渡态和中间体的各极值点结构,绘制了布洛芬分子手性转变路径反应势能面,分析了各极值点的几何和电子结构特性.结果表明:布洛芬实现从S型到R型手性转变的反应路径有两条.路径1包括三个过渡态和两个中间体,路径2包括四个过渡态和三个中间体.反应路径上最大的能垒是73.54 Kcal/mol,来源于手性碳上的氢向羧基上的氧转移.这一研究为进一步实现一些有重要应用价值的点手性分子手性转变反应调控提供了理论参考.
手性; 布洛芬; 密度泛函理论; 过渡态
1 引 言
布洛芬,分子式:C13H18O2,具有抗炎、镇痛、解热作用.用于治疗风湿性关节炎、类风湿性关节炎、骨关节炎、强直性脊椎炎和神经炎等.关于布洛芬的研究已有很多相关的报道.Subhash Bhatia等人对外消旋布洛芬的动力学拆分做了模拟和实验的研究[1].肖方青等人的研究发现,布洛芬的药理活性主要来自右旋体,右旋布洛芬在疗效、安全性和药动学特性方面都优于外消旋布洛芬[2].林文辉报道,右旋体的活性是左旋体的160倍,外消旋体的1.6倍,在体内可以实现左旋体向右旋体的缓慢转变[3].但目前对布洛芬的手性转变机制却未见报道.
一般我们得到的都是外消旋体,导致市售的产品多数为消旋体[4].因此,寻找一个更有效的从已有的布洛芬外消旋体药物,将其中的“劣构体”转化成有效的单一异构体“优构体”的途径变得尤为重要.本工作希望,通过对孤立条件下布洛芬分子手性转变路径及反应所要克服的能垒的研究,得到布洛芬分子手性转变的反应机制.为布洛芬在体内缓慢转变的理论研究做必要的准备工作,为实验上获得光学纯的布洛芬从理论提供一条新途径.
2 研究与计算方法
基于密度泛函理论的B3LYP[5,6]方法,采用双分裂价基,对C、O等原子加了d 极化函数,对H原子加了p极化函数.即采用6-31+g(d,p)基组,进行单重态势能面上的极小值、红外振动频率及前线分子轨道的理论计算.把S型布洛芬分子做为反应物,研究寻找到产物R型布洛芬分子的过渡态[7-9]及中间体,并对包括过渡态在内的极值点的前线分子轨道进行了分析以获得分子的键特性.将反应物、过渡态、中间体、产物等各极值点连接起来确定反应路径.
为验证过渡态的可靠性,对过渡态进行了内禀反应坐标IRC分析[10-13].文中理论计算及分子结构等图形由Gaussian03/GaussView3.0软件程序完成.
3 结果与讨论
3.1 布洛芬分子手性对映体的结构与分析
图1是在B3LYP/6-31+g(d,p)水平上获得的布洛芬手性对映体的结构.已有报道,氢转移过程对于点手性分子对映体转变是最佳的反应途径[14]. 从这对对映体的几何结构可以看出,要实现这对对映体从S型到R型的手性转变,必须实现32H从纸面外的位置迁移到12C的纸面里的位置.依据基本的分子结构理论和我们的研究经验,推测手性转变的过程是:33H先转移到31O上,然后32H转移到30O上,最后32H或33H再转移到12C的另一侧.过程中伴随着:4C-12C-17C、11C-13C-14C-15C碳骨架的异构; 11C、14C、15C、16C等基团的旋转异构:C环平面绕11C、1C、4C与12C所在轴的旋转异构; 32H-12C-16C键角对称改变的异构.从而完成对映体的手性转变过程.

图1 在B3LYP/6-31+g(d,p)水平上的S型与R型布洛芬分子的几何结构Fig.1 The geometries of S-type and R-type ibuprofen molecule in B3LYP/6-31+g(d,p) level

图2 在B3LYP/6-31+g(d,p)水平上的INT1的几何结构(a)与前线分子轨道(b)和(c)Fig.2 The geometry (a), frontier molecular orbitals (b) and (c)of INT1 in B3LYP/6-31+g(d,p) level

图3 在B3LYP/6-31+g(d,p)水平上的INT2的几何结构(a)与前线分子轨道(b)和(c)Fig.3 The geometry (a), frontier molecular orbitals (b) and (c) of INT2 in B3LYP/6-31+g(d,p) level
3.2 布洛芬分子手性转变路径中间体的计算及过渡态的探索
根据前面的分析得到布洛芬分子手性转变过程:先是S型布洛芬经过过渡态TS1实现33H到31O上的转移,形成中间体INT1.然后经过过渡态TS2,32H转移到30O上,形成中间体INT2.接着是33H或32H转移到12C的另一侧.前者是INT2经过渡态TS3直接形成R型布洛芬分子.后者是INT2经过渡态TS4行成中间体INT4, INT4经过过渡态TS5实现33H迁回到30O上的过程,得到产物R型布洛芬异构体.
3.2.1 布洛芬分子手性转变路径的中间体结构特性
在B3LYP/6-31+g(d,p)水平上对各中间体进行几何结构优化、计算单重态的最低单点能、红外振动频率及前线分子轨道.
INT1的结构如图2无虚频.前线分子轨道见图2,主要来源于骨架苯环C与O原子的p电子的贡献,骨架原子苯环C间、1C与11C间、12C与16C之间展现了π键效应,其它原子的p电子展现了局域特性,32H和33H与相邻骨架原子展现的非键特性.
INT2的结构如图3无虚频.前线分子轨道见图3,来源于除去14C与15C以外的骨架原子的p电子的贡献,展现了一种π成键效应,17C原子的p电子展现了局域特性.LUMO轨道显示32H与33H与O原子的非键特性.
第一种情况是INT2的33H迁移到12C生成的产物优化得到结构如图4.对图1与图4的R型布洛芬分子的键长、键角、二面角数据进行比较,数据都是一样的(相差都在0.010到0.10之间,甚至更小).证明得到的结构确实是S型布洛芬分子的手性对映体R.
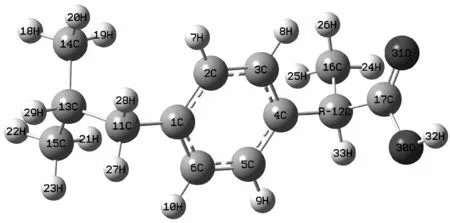
图4 R型布洛芬分子的几何结构Fig.4 The geometry of R-type ibuprofen molecule

图5 INT4的几何结构Fig.5 The geometry of INT4
第二种情形是32H迁移到12C生成中间体产物INT4,最后33H再迁回到30O形成产物R.INT4的结构如图5,其能量与INT1相同.前线分子轨道见图6,主要来源于除去11C、14C与15C以外的骨架原子的p电子的贡献,并展现了一种π成键效应,30O原子的p电子展现了局域特性.LOMO轨道明显显示33H与O原子的非键特性.
INT4的33H经TS5再迁移回到30O的生成物优化后构型同图1的R型布洛芬.对键长、键角、二面角数据进行了比较,数据都是一样的(相互差别都在0.010到0.10之间,甚至更小).各个稳定点的能量见表1.
3.2.2 布洛芬分子手性转变过程的过渡态结构特性
在B3LYP/6-31+g(d,p)水平上,对过渡态TS1、TS2、TS3、TS4与TS5进行了的探索.得到了它们的几何构型、前线分子轨道及虚频下的振动模式,分别如图7、8、9、10和图11所示(TS2和TS4的前线分子轨道分别与TS3和TS5的雷同,这里从略).
沿着虚频下相关原子的振动方向微调结构,进行结构优化的结果分别为每个过渡态对应的反应物与产物,证明了诸过渡态是正确的.
这五个过渡态的前线分子轨道具有共性.篇幅所限,只给出具有代表性的TS1、TS3和TS5的前线分子轨道,见图7、9、11.TS1的HOMO轨道是30O和31O原子的p电子与33H原子的S电子贡献了这个具有明显反键特征的轨道,而其LUMO轨道是31O原子的p电子与33H原子的S电子贡献了这个具有成键特征的轨道.TS3和TS5的前线分子轨道也体现了过渡态的非键特性,具体的分析从略.
在B3LYP/6-31+g(d,p)水平上对过渡态进行全优化后的能量及过渡态的虚频见表1.

图6 在B3LYP/6-31+g(d,p)水平上的INT4的前线分子轨道(a)和(b)Fig.6 The frontier molecular orbitals (a) and (b) of INT4 in B3LYP/6-31+g(d,p) level

图7 TS1几何结构、虚频的振动模式(a)及前线分子轨道(b)和(c) Fig.7 The geometry,vibration modes (a) under imaginary frequency and frontier molecular orbitals (b) and (c) of TS1

图8 TS2的几何结构及虚频下的振动模式Fig. 8 The geometry and vibration modes under imaginary frequency of TS2
3.3 布洛芬分子手性转变的路径
综合前面的分析与研究,确认布洛芬分子从S型到R型的手性转变有两个路径.路径1为:S→TS1→INT1→TS2→INT2→TS3→R,过程局域极小点的几何结构见图1至图4,过渡态的结构见图7、8、和9.路径2为:S→TS1→INT1→TS2→INT2→TS4→INT4→TS5→R,过程局域极小点的几何结构见图1、2、3、5,过渡态的结构见图7、8、10和11.
为形象描述反应过程中各中间体、过渡态和反应物与产物的能量变化,每步反应的能垒,并使反应过程更加清晰.依据表1的数据,绘制了手性转变过程的路径1与路径2所对应的势能面示意图,见图12.

图9 TS3几何结构虚频的振动模式(a)及前线分子轨道(b)和(c)Fig.9 The geometry, vibration modes (a) under imaginary frequency and frontier molecular orbitals (b) and (c) of TS3

图10 TS4的几何结构及虚频下的振动Fig. 10 The geometry and vibration modes under imaginary frequency of TS4
从图12可以看出,对于路径1:S需跨越能垒34.53 Kcal/mol,经TS1形成INT1;INT1需跨过73.54Kcal/mol的能垒,经TS2转变到INT2; INT2经TS3转变到R,需跨过51.67 Kcal/mol的能垒.对于路径2:S到到INT2的过程与路径1的相同,而后INT2跨过50.66 Kcal/mol的能垒经TS4形成INT4, 最后INT4跨过33.65 Kcal/mol的能垒,经过TS5转变到R型布洛芬.表2和3给出了反应路径1和2中反应物、中间体、过渡态及产物结构的主要几何参数.
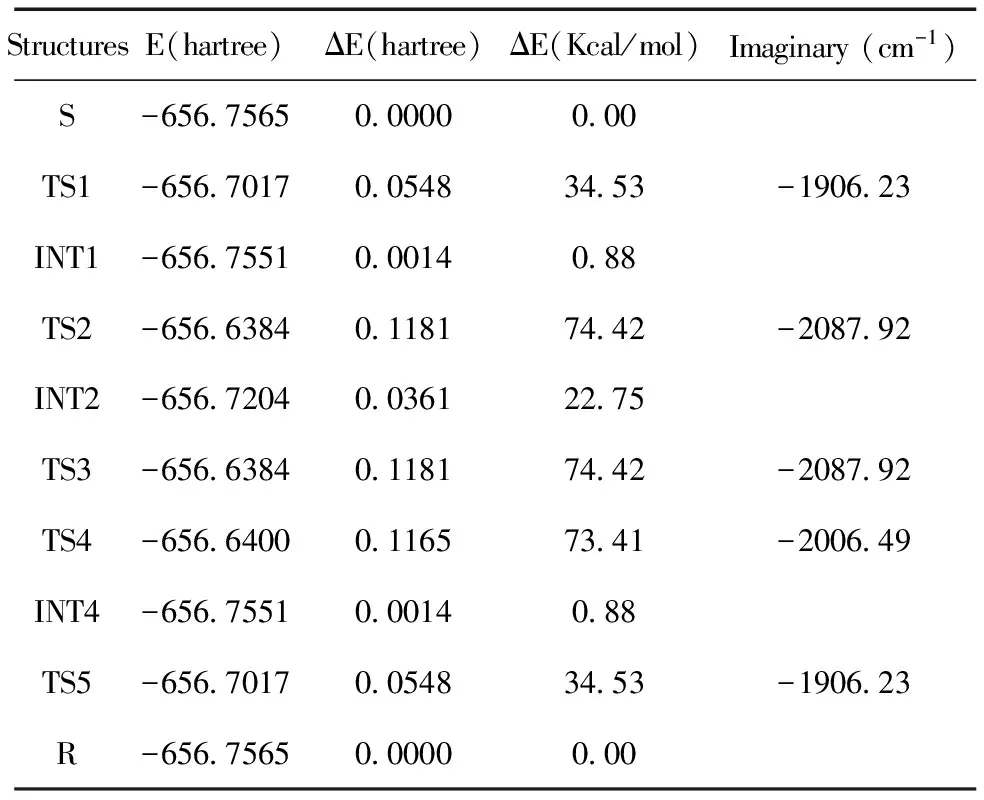
表1 在B3LYP/6-31+g(d,p)理论水平上计算得到的反应路径中,各稳定点及过渡态的能量,过渡态、中间体、反应物及产物的能量差及过渡态的虚频

图11 TS5几何结构、虚频的振动模式(a)及前线分子轨道(b)和(c)Fig.11 The geometry, vibration modes (a) under imaginary frequency and frontier molecular orbitals (b) and (c) of TS5
表2 的数据显示了,S型布洛芬分子经过过渡态TS1、TS2、TS3分别达到中间体INT1、INT2和R型布洛芬分子过程中,反应物、中间体、过渡态及产物结构的主要二面角的变化, 说明了S型布洛芬分子经路径1完成了手性转变,异构化为R型布洛芬分子.
表3 的数据,显示了S型布洛芬分子经过渡态TS1、TS2、TS4、TS5分别达到中间体INT1、INT2、INT4和R型布洛芬分子过程中,反应物、中间体、过渡态及产物结构的主要二面角的变化过程,说明了S型布洛芬分子经路径2完成了手性转变,异构化为R型布洛芬分子.

图12 在B3LYP/6-31+g(d,p)水平上手性转变过程的势能面示意图Fig.12 The schematic of potential energy surface on chiral transition process in B3LYP/6-31+g(d,p) level

StructuresαβγδθωS-122 88-60 398-117 39126 26127 12179 46TS1-123 45-179 20-119 79126 63127 18179 21INT1-125 72-179 53-119 46126 66127 12179 49TS2-150 15-173 12-118 73126 49127 31179 72INT2-178 17-178 98-120 47126 51127 19179 89TS3150 15173 12121 11-126 49-127 32-179 72R122 8860 398117 39-126 62-127 12-179 45

表3 在B3LYP/6-31+g(d,p)理论水平上计算得到反应路径2中各稳定点及过渡态的主要几何参数
3.4 布洛芬分子手性转变过程过渡态的IRC分析
为进一步验证过渡态的可靠性,对过渡态在B3LYP/6-31+g(d,p)理论水平上进行了IRC计算,结果如图13、14、15、16所示,最高点对应的是过渡态,TS5与TS1的IRC基本相同,从略.
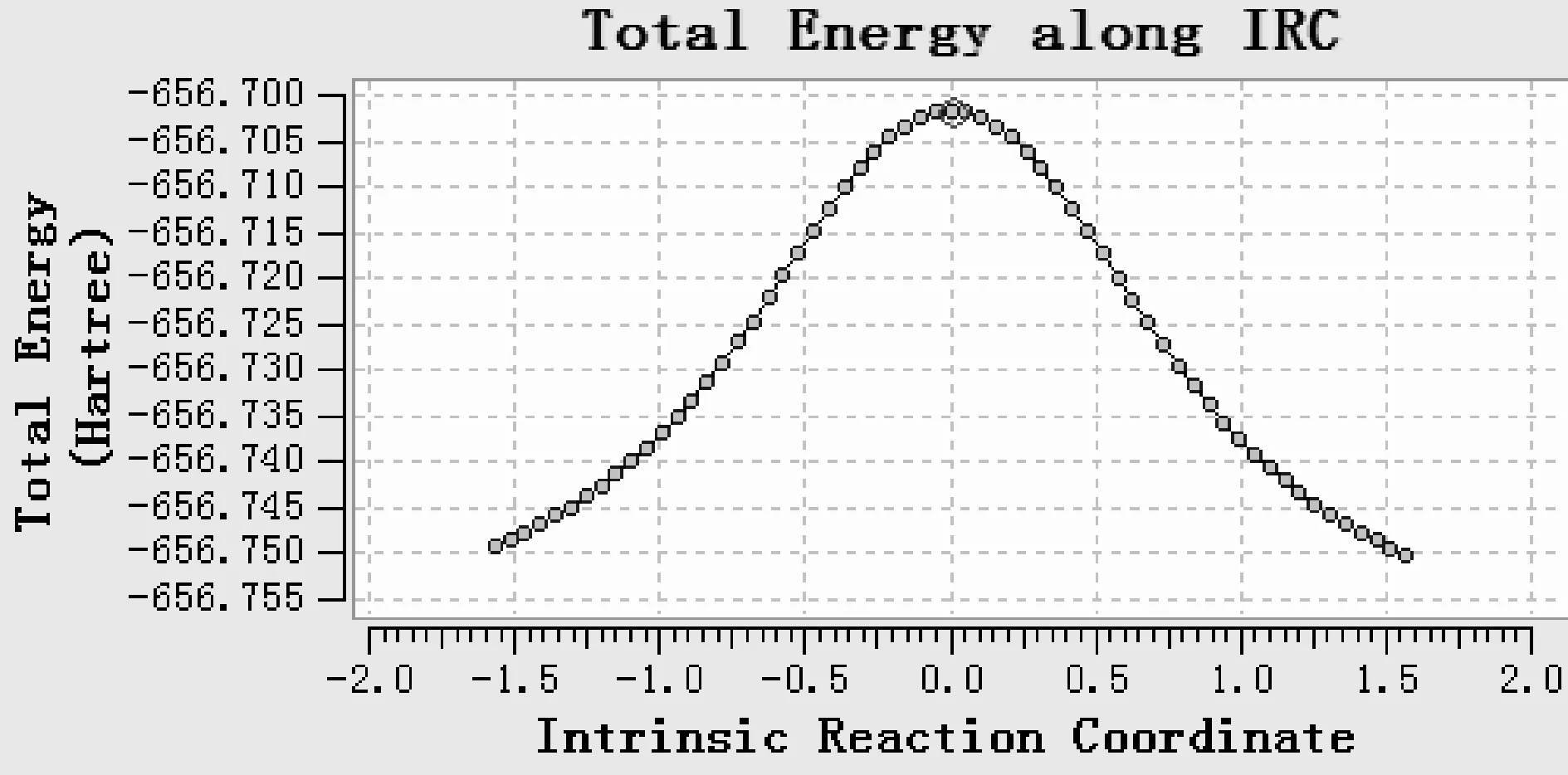
图13 对过渡态TS1进行的IRC分析最低点:右侧指向S,左侧指向INT1Fig.13 IRC analysis of transitive state TS1.The lowest point: the right side points to S,the left side points to INT1
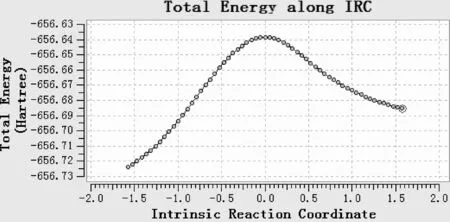
图14 对过渡态TS2进行的IRC分析最低点:左侧指向INT1,右侧是指向INT2Fig.14 IRC analysis of transitive state TS2.The lowest point: the left side points to INT1,the right side points to INT2

图15 对过渡态TS3进行的IRC分析最低点:右侧指向INT2,左侧指向RFig.15 IRC analysis of transitive state TS3.The lowest point: the right side points to INT2, the left side points to R

图16 对过渡态TS4进行的IRC分析最低点:右侧指向INT2,左侧指向INT4Fig.16 IRC analysis of transitive state TS4.The lowest point: the right side points to INT2, the left side points to INT4
对IRC路径两端对应的反应物与产物进行结构优化,验证了诸过渡态的可靠性.在TS1的IRC路径上取了几个有代表性的点的结构,得到图17,清晰地说明了S变为INT1的过渡态TS1的可靠性.在其它IRC路径上分子结构的异构化过程,也同样说明了对应的过渡态的可靠性.由于篇幅所限这里从略.

图17 S型对映体转变为INT1的简明过程:A→B→C→D→E→F→G→HFig.17 The brief conversion from S-type enantiomer into INT1: A→B→C→D→E →F → G→H
3 结 语
使用密度泛函的B3LYP方法,采用6-31+g(d,p)基组,计算研究了布洛芬分子从S型到R型的转变过程.结果表明,此反应的路径有两个:一是S →TS1→INT1→TS2→INT2→TS3→R.此过程要经过三个过渡态和二个中间体,需要跨过的能垒分别是34.53Kcal/mol、73.54 Kcal/mol和51.67 Kcal/mol.二是S→TS1→INT1→TS2→INT2→TS4→INT4→TS5→R.此路径上,从S到INT2的过程同路径1.而后是跨越50.66 Kcal/mol的能垒,经TS4转变到INT4.最后再跨过33.65 Kcal/mol的能垒,经TS5转变到R,实现手性对映体转变.由于这两个路径上都有比较高的能垒,所以布洛芬的结构在通常情况下是稳定的,其手性转变过程需要在一定的外界条件干预下才能实现.对过渡态前线分子轨道的分析与研究和对过渡态进行的IRC计算与研究进一步表明,得到的诸过渡态是可靠的.本研究对获得光学纯的包括布洛芬在内的点手性分子对映体结构及其在生物体内的手性转变机制等研究工作具有一定的参考意义.
[1] Subhash B, Wei S L. Enzymatic membrane reactor for the kinetic resolution of racemic ibuprofen ester: modeling and experimental studies[J].ChemicalEngineeringScience, 2004, 59: 5061.
[2] Xiao F Q. The preparation of dexibuprofen[J].ChineseJournalofPharmaceuticals, 2000, 31(11): 486(in Chinese)[肖方清.右旋布洛芬的制备[J].中国医药工业杂志, 2000, 31(11): 486]
[3] Lin W H.Kineticsofchiraldrugibuprofeninvivodrug[D]. Shenyang Pharmaceutical University, 2004(in Chinese)[林文辉.手性药物布洛芬的体内药物动力学研究[D].沈阳药科大学, 2004]
[4] Zhao Y H.Molecularbiologytutorial[M]. Beijing: Science Press, 2011(in Chinese)[赵亚华.分子生物学教程[M].北京: 科学出版社, 2011]
[5] Becke A D. Density-functional thermochemistry. III. The role of exact xchange[J].J.Chem.Phys., 1993, 98: 5648.
[6] Xu G X.QuantumChemistry[M]. Beijing: Science Press, 1999(in Chinese)[徐光宪.量子化学[M].北京: 科学出版社, 1999]
[7] Eyring H. The activated complex and the absolute rate of chemical reaction[J].ChemicalReviews, 1935, 17(1): 65.
[8] Garrett B C,Truhlar D G. Generalized transition state theory.Classical mechanical theory and applications to collinear reactions of hydrogen molecules [J].JournalofPhysicalChemistry, 1979, 83(8): 1052.
[9] Garrett B C, Truhlar D G. Criterion of minimum state density in the transition state theory of bimolecular reactions[J].TheJournalofChemicalPhysics, 1979, 70(4): 1593.
[10] Fukui K. Formulation of the reaction coordinate[J].TheJournalofPhysicalChemistry, 1970, 74(23): 4161.
[11] Gonzalez C, Schlegel H. An improved algorithm for reaction path following[J].TheJournalofChemicalPhysics, 1989, 90: 2154.
[12] Gonzalez C, Schlegel H. Reaction path following in mass-weighted internal coordinates[J].JournalofPhysicalChemistry, 1990, 94(14): 5523.
[13] Ishida K, Morokuma K, Komornicki A. The intrinsic reaction coordinate. An ab initio calculation for HNC→HCN and H-+ CH4→CH4+ H-[J].TheJournalofChemicalPhysics, 1977, 66: 2153.
[14] Tian C J. Enantiomerization mechanism of thalidomied and the role of water and hydroxide ions[J].Chem.Eur.J., 2012, 18: 14305.
The theoretical research on the chiral transition of ibuprofen molecules under isolated conditions
ZOU Xiao-Wei1, MEI Ze-Min2, WANG Li-Ping3, TONG Hua3, YU Tian-Rong3, WANG Zuo-Cheng3,4
(1.Mathematical Research Department, Haikou Economies College, Haikou 570100, China; 2.Chemistry Department, Baicheng Normal College, Baicheng 137000, China; 3.Physics Department, Baicheng Normal College, Baicheng 137000, China; 4. Institute of Atomic and Molecular Physics, Jilin University, Changchun 130012, China)
In this article, we do a research on the chiral shift process of the isolated alpha alanine molecule using the basis set of 6-31+g(d,p), which is based on density functional theory B3LYP. Further more, the chiral transition path reaction potential energy surface of ibuprofen molecule is drawn by looking for the extreme value point structure including the transition state and intermediate. Finally, the geometry and electronic structure properties of extreme value point are also analyzed. The results show that there are two achieve reaction paths of ibuprofen from S-type to R-type. Path 1 consists of three transition states and two intermediate states. Path 2 includes four transition states and three intermediate states. On the reaction path, the greatest barrier which is from the transfer of hydrogen in chiral carbon to oxygen in carboxyl, is 73.54 Kcal / mol. The research provides a theoretical reference to further realize some important application value over the chiral transition reaction control of point chiral molecule.
Chiral; Ibuprofen; Density functional theory; Transition state
103969/j.issn.1000-0364.2015.02.001
2013-03-09
吉林省科技发展计划资助项目自然科学基金(20130101131JC);白城师范学院科技计划重点项目(2013第A2号)
邹晓威(1980—),女,讲师,硕士,研究方向为原子与分子物理.E-mail: 330453913@qq.com
王佐成.E-mail: wangzc188@163.com
O641.12+1
A
1000-0364(2015)02-0173-08
