16S rDNA测序技术在婴儿肠道微生态研究中的应用*
2015-03-15马丽亚王辉林卢光进
马丽亚,王辉林,陈 睿,张 敏,黄 艳,卢光进△
(深圳市宝安区妇幼保健院:1.新生儿科;2.中心实验室 518133)
·论 著·
16S rDNA测序技术在婴儿肠道微生态研究中的应用*
马丽亚1,王辉林2,陈 睿1,张 敏1,黄 艳1,卢光进1△
(深圳市宝安区妇幼保健院:1.新生儿科;2.中心实验室 518133)
目的 探讨16S rDNA测序技术在新生儿、婴儿肠道微生态研究中的应用。方法 于生后3天、1月、6月、1岁时收集2例健康婴儿粪便标本共8份,提取细菌总DNA,以Illumina Hiseq 2000为测序平台,采用新一代高通量16S rDNA宏基因组测序技术对V6可变区测序,并进行生物信息分析(物种分类和丰度分析;多样性分析)。结果 8份样品共产生原始测序数据为1 027.47 Mbp,Unique tags序列数量均值为58630,OTU数量63~209;优势菌门为Proteobacteria和Firmicutes; 在科水平,>1%的物种1个月之内2~4种,6月后达7~10种;1号婴儿一直以Enterobacteriaceae占优势,2号婴儿优势菌群包括Enterobacteriaceae、Lachnospiraceae、Streptococcaceae和Bacteroidaceae;4个时间点的npShannon和Simpson指数分别为1.17、1.29、2.16、2.51和0.43、0.40、0.26、0.14。结论 16S rDNA测序技术能满足新生儿、婴儿肠道微生态研究需求;新生儿、婴儿粪便中含丰富细菌基因组;细菌物种丰度及分类存在个体差异;从出生到1岁,婴儿肠道菌群结构趋向复杂和多样。
肠道菌群; 测序; 宏基因组; 婴儿
寄居于人体内(主要是肠道)的细菌基因组是人体自身基因组的100多倍[1-2],被称为人类的“第二基因组”[3],肠道菌群对营养物质的消化、吸收、对抗病菌及免疫调节都发挥着重要作用[4]。新生儿、婴儿期是人体肠道菌群定植并发育的重要阶段,此期肠道菌群的失衡可能影响远期甚至成年后的健康[1]。因而,了解新生儿、婴儿期肠道菌群的发展模式及特点对研究相关疾病、改善人口质量有重要意义。
对肠道微生态的研究以往多采用培养或传统分子生物学(如PCR、变性梯度凝胶电泳)方法,但它们对于研究复杂的肠道菌群有明显的局限性,而宏基因组学研究则可以避免上述方法的缺陷[5]。最近,有关中国婴儿肠道宏基因组的研究初见报道[6],但缺乏连续性研究和新生儿资料,本研究拟采用以Illumia为测序平台的16S rDNA新一代高通量测序技术对2例健康婴儿从新生儿期至1岁以内的粪便细菌基因组进行连续分析,探讨该方法在新生儿、婴儿肠道微生态研究中的应用,为以后大规模研究中国婴儿肠道菌群分布特征及婴儿肠道微生态相关疾病提供参照。
1 资料与方法
1.1 一般资料 选择2例间隔1周出生于深圳市宝安区妇幼保健院同一病区的健康足月新生儿为研究对象,追踪至1岁。2例婴儿胎龄分别为38周、39周,均为女孩,剖宫产出生,无胎膜早破,无产时窒息,母亲术后预防性应用头孢唑啉2剂,生后一直混合喂养至1岁。本课题经本院伦理委员会同意并获得了婴儿父母的知情同意。
1.2 粪便标本的留取及提取细菌总DNA 生后3 d(时间点标注为a)、1月(标注为b)、6月(标注为c)和1岁(标注为d)时用无菌标本盒留粪便不少于10 g,1 h内冷冻于-70 ℃,留取粪便前1周婴儿不使用抗生素、益生菌,如有疾病,可暂缓1周留取。在本院中心实验室采用德国Qiagen公司生产的QIAamp DNA Stool Mini Kit试剂盒提取粪便细菌总DNA,保存于-70 ℃,具体步骤由专人按说明书操作。DNA标本于冷藏条件下送至深圳华大基因科技服务有限公司进行测序及生物信息分析。
1.3 16S rDNA测序 用细菌通用引物对粪便DNA样品进行16S rRNA基因V6区PCR扩增,然后回收电泳主要的DNA片段后构建文库,制备cluster;应用Hiseq 2000测序仪(Illumina公司,美国)对扩增的16S rDNA高变区序列进行测序,测序区域为V6区,长度为100 bp,测序类型为101 PE测序。
1.4 生物信息分析
1.4.1 物种分类与丰度分析 对原始测序数据处理后获取clean data,拼接成tags;利用Mothur (version 1.27.0)软件包对tags序列进行了去冗余处理,从中挑选出unique tags序列并进行物种注释,聚类成Operational Taxonomic Units (OTU),Tags和OTU的数量初步说明了该样品是否具有丰富的物种;然后通过OTU 注释(基于包含的tags的物种注释结果)完成物种组成和分类分析,可在界、门、纲、目、科、属、种水平,将每个注释上的物种或OTU在不同样品中的序列数进行整理。
1.4.2 多样性分析 利用Mothur(version 1.27.0)软件计算针对单个样品物种多样性分析的Alpha多样性值,包括chao1值、ACE值、Shannon、npShannon以及Simpson指数[7],前面4个指标数值越大,最后一个指标越小,说明样品中的物种越丰富。
2 结 果
2.1 样品测序数据 8份粪便细菌总DNA样品共产生原始测序数据为1 027.47 Mbp,每份样品平均产生124.53 Mbp的clean data序列,每一样品信息的利用率都在94%以上,出生仅3 d时已含丰富的细菌基因组。
2.2 物种分类和丰度分析 各样品检测的Tags数量平均为69 840,Unique tags序列数量均值为58 630;OTU数量63~209,随着年龄的增长而增加,3 d、1月、6月和1岁时平均为67、86、149和176,表明婴儿粪便样品中微生物丰度随时间而增加。在门(Phylum)水平,20个菌门被检出细菌基因组,但主要分布于变形菌门(Proteobacteria)和厚壁菌门(Firmicutes)2个菌门,2例婴儿粪便中比例差异有统计学意义。另外,2号婴儿的拟杆菌门(Bacteroidetes)在1岁时上升至22%。见表1。
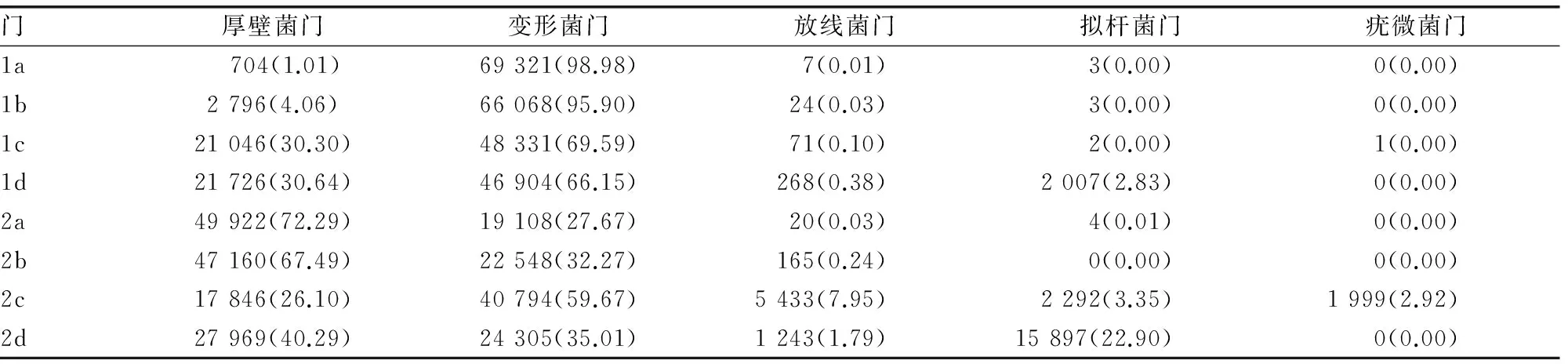
表1 门水平主要细菌占比情况表[tags数量及百分比,n(%)]
注:a、b、c、d分别代表3 d、1月、6月、1岁4个时间点。
本研究中最佳的分类水平为科,每个样品均有80%以上的Tags在该水平上得到分类。在科水平,2例婴儿肠道菌群中超过1%的物种1个月之内仅2~4种,6月后达7~10种。1号婴儿一直以Proteobacteria下的肠杆菌科(Enterobacteriaceae)占优势,其他包括Firmicutes下的梭菌科(Clostridiaceae)、毛螺旋菌科(Lachnospiraceae)等,双歧杆菌科(Bifidobacteriaceae)一直未超过1%。2号婴儿优势菌群包括Enterobacteriaceae以及Firmicutes下的毛螺旋菌科(Lachnospiraceae)、链球菌科(Streptococcaceae)和Bacteroidetes下的拟杆菌科(Bacteroidaceae),双歧杆菌科最高时也仅7.3%。
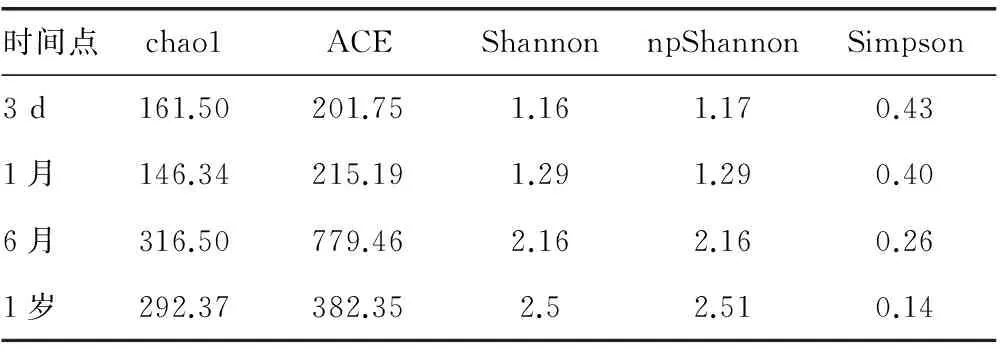
表2 样品在0.03距离下的不同时间点Alpha丰富 程度各指数均值
2.3 样品多样性分析 3 d、1月、6月及1岁时chao1值、ACE值、Shannon、npShannon值越来越大,而Simpson指数越来越小,提示粪便中菌群多样性随着时间而增加,物种越来越丰富。见表2。
3 讨 论
高通量测序技术又称新一代或第二代测序技术,以边合成边测序为原理,可以一次性对几十万至几百万条DNA分子进行序列测定,而以Illumina为测序平台的测序技术在研究人类肠道宏基因组方面发挥着举足轻重的作用[2-3,6,8]。16S rDNA是细菌进化以及分类研究最常用的靶分子,16S rDNA宏基因测序技术不需进行克隆筛选,测序的通量高,获得的数据量大,周期短,与传统的培养鉴定和PCR、变性梯度凝胶电泳、原位杂交等分子生物学方法相比,能更全面反映微生物群体的物种组成、分布及丰度信息。本研究对2例剖宫产出生、混合喂养的健康婴儿粪便标本于1岁之内连续进行16S rDNA的V6高变区新一代高通量测序,8份粪便细菌总DNA样品共产生原始测序数据为1 027.47 Mbp,每份样品平均产生124.53 Mbp的clean data序列。Barrett等[9]对10例早产儿生后2周和4周的粪便标本进行16S rDNA的V4区进行测序分析(Genome Sequencer FLX测序平台,Roche Diagnostics Ltd,West Sussex,UK),20份标本共产生529 102 bp原始序列和452 863 bp clean data 序列,本研究测序数据多于Barrett等对于早产儿、新生儿的报道,原因可能与测序平台、婴儿胎龄、日龄不同有关,同时也表明以Illumina为测序平台的16S rDNA测序技术能够满足新生儿、婴儿肠道微生态研究的需求。
影响新生儿肠道菌群定植的因素包括分娩方式[10]和喂养方式[6]等,例如,剖宫产出生的婴儿其肠道菌群多样性减少、益生菌拟杆菌门定植延迟[10];母亲肠道是阴道分娩婴儿肠道菌群定植的发源地,而母乳中的细菌以及通过吸吮接触乳头周围的皮肤是母乳喂养婴儿肠道菌群的重要来源[1];母亲围生期使用抗菌药物可改变其婴儿肠道菌群结构[11]。本研究中 2例婴儿虽然出生环境及围产期因素相似(同一产区、均为剖宫产出生、混合喂养、母亲产时均有预防性抗菌药物应用),但粪便中的菌群分布却有明显的差异,说明除了以上常见影响因素外,尚有其他影响婴儿肠道细菌定植的因素,如地域[11],家庭环境(如宠物)或兄弟姐妹的影响[12]等,从而造成婴儿肠道宏基因组独特的个体特征,但这些结论还需要大标本、多地域人群的连续研究来证实。另外,常见的益生菌双歧杆菌科在2例婴儿肠道中的比例均较低,与Fan等[6]的研究类似。
不同于Fan等[6]仅一个时间点的研究,本研究表明,新生儿出生仅3 d时肠道中已含有丰富的细菌基因组,从出生到1岁,婴儿粪便中细菌丰度及多样性随时间而增加,提示随着婴儿饮食结构的复杂和个体的发育,以及活动范围的扩展,婴儿肠道菌群结构趋向复杂和多样。但值得注意的是,出生不久即定植的优势菌群,1岁以内可能会一直存在,例如1号婴儿肠道中一直以肠杆菌科占优势,而2号婴儿肠道中自1月时出现的毛螺旋菌科此后亦一直为优势菌群,说明生命早期的因素可能会影响1岁以内甚至儿童、成人期的肠道菌群结构。
本研究提示,16S rDNA测序技术能满足新生儿、婴儿肠道微生态研究需求;婴儿粪便中含丰富细菌基因组;细菌物种丰度及分类存在个体差异;从出生到1岁,婴儿肠道菌群结构趋向复杂和多样;本研究将为未来新生儿、婴儿的大样本、分组研究提供线索。
[1]Matamoros S,Gras-Leguen C,Le Vacon F,et al.Development of intestinal microbiota in infants and its impact on health[J].Trends Microbiol,2013,21(4):167-173.
[2]Qin J,Li R,Raes J,et al.A human gut microbial gene catalogue established by metagenomic sequencing[J].Nature,2010,464(7285):59-65.
[3]Song S,Jarvie T,Hattori M.Our second genome-human metagenome:how next-generation sequencer changes our life through microbiology[J].Adv Microb Physiol,2013,62:119-144.
[4]Di Mauro A,Neu J,Riezzo G,et al.Gastrointestinal function development and microbiota[J].Ital J Pediatr,2013,39:15-18.
[5]丁贤,殷波,李慧贤,等.宏基因组学在微生物活性物质筛选中的应用[J].中国微生态学杂志,2010,22(6):565-568
[6]Fan W,Huo G,Li X,et al.Impact of diet in shaping gut microbiota revealed by a comparative study in infants during the six months of life[J].J Microbiol Biotechnol,2014,24(2):133-143.
[7]Kemp PF,Aller JY.Bacterial diversity in aquatic and other environments:what 16S rDNA libraries can tell us[J].FEMS Microbiol Ecol,2004,47(2):161-177.
[8]Qin J,Li Y,Cai Z,et al.A metagenome-wide association study of gut microbiota in type 2 diabetes[J].Nature,2012,490(7418):55-60.
[9]Barrett E,Kerr C,Murphy K,et al.The individual-specific and diverse nature of the preterm infant microbiota[J].Arch Dis Child Fetal Neonatal Ed,2013,98(4):F334-F340.
[10]Jakobsson HE,Abrahamsson TR,Jenmalm MC,et al.Decreased gut microbiota diversity,delayed Bacteroidetes colonisation and reduced Th1 responses in infants delivered by Caesarean section[J].Gut,2014,63(4):559-566.
[11]Fallani M,Young D,Scott J,et al.Intestinal microbiota of 6-week-old infants across Europe:geographic influence beyond delivery mode,breast-feeding,and antibiotics[J].J Pediatr Gastroenterol Nutr,2010,51(1):77-84.
[12]Azad MB,Konya T,Maughan H,et al.The gut microbiome and the hygiene hypothesis of allergic disease.Impact of pets and siblings on infant gut microbiota[J].Ann Am Thorac Soc,2014,11(Suppl 1):S73-S76.
Application of 16S rDNA sequencing technique in infantile intestinal microecological research*
MALi-ya1,WANGHui-lin2,CHENRui1,ZHANGMin1,HUANGYan1,LUGuang-jin1
(1.DepartmentofNeonatology;2.CentralLaboratory,ShenzhenBao′anDistrictMaternalandChildHealthCareHospital,Shenzhen,Guangdong518133,China)
Objective To investigate the application of 16S rDNA sequencing technique in studying intestinal microecology of neonates and infants.Methods Eight fecal samples were collected on 3 d,1 month,6 months and 1 year in 2 healthy infants.Total bacterial DNAs were extracted and submitted the high throughout 16S rDNA sequencing on the V6 viable region by using Illumia genome analyzer Hiseq 2000 (101 bp pair-end sequencing strategy).The 16S rDNA tags and operational taxonomic units (OTU) were then obtained from the sequences.The bioinformatic analysis including analysis of taxonomy,abundance and alpha diversity were performed.Results Total 1 027.47 Mbp raw data were produced.The mean unique tags number was 58630.The OTU numbers ranged from 36 to 308.The bacterial families more than 1% were increased from 2-4 species per sample before 1 month to 7-10 species after 6 months.Enterobacteriaceae was always the predominant family in No.1 infant through the first year,while in No.2 infant the predominant groups included Enterobacteriaceae,Lachnospiraceae,Streptococcacea and Bacteroidaceae.The mean npShannon and Simpson indexes at 4 time points were 1.17,1.29,2.16,2.51 and 0.43,0.40,0.26,0.14 respectively.Conclusion 16S rDNA sequencing could meet the need of studying intestinal microecology in neonates and infants;neonatal and infantile feces contains abounding bacterial genomes;the individual differences exist in intestinal bacterial abundance and composition;the structure of gut bacterial floras trend to complexity and diversity from birth to 1 year old.
intestinal microbiota; sequencing; metagenomics; infant
广东省深圳市科技计划项目(201102030)。
马丽亚,女,主任医师,硕士,研究方向包括新生儿营养与肠道微生态、新生儿肺损伤。
△通讯作者,E-mail:lugj1111@aliyun.com。
10.3969/j.issn.1672-9455.2015.02.011
A
1672-9455(2015)02-0166-03
2014-05-21
2014-10-30)
