焙烧与急冷过程中钾元素组成及其形态转变因素探析
2015-03-10乌红绪李卫昌
乌红绪,李卫昌
(金堆城钼业股份有限公司,陕西 西安 710077)
0 引言
金属钼除在钢铁、有色、化工等工业领域有着广泛的用途外,低钾钼粉在超大集成电路、高清晰度电视、LCD 液晶显示器、靶材等电子工业应用领域不断拓宽[1]。
低钾钼粉是通过钼采矿、选矿、焙烧直到钼化工、钼加工产业链生产而制成的,其中由多膛炉焙烧生产的产品中钾的含量以及组成形态的变化对后续钼化工过程起着至关重要的作用,因而必须合理地优化控制多膛炉系统焙烧与急冷生产工艺,并有效地降低焙烧钼精矿钾含量和改变其组成形态,最大限度地生产优质低钾高溶焙烧钼精矿产品,以便更好地为后续钼化工创造有利的生产作业条件[2]。
1 影响焙烧与急冷过程中钾元素组成及其形态转变因素探析
生产实践表明,多膛炉生产的焙烧钼精矿产品中有3 种含钾化合物:一是水溶性含钾化合物,在ADM 工艺水浸出过程中除去;二是水不溶且NH4OH不溶的含钾化合物,ADM 工艺NH3溶解过程过滤步骤中去除;三是水不溶但NH4OH 可溶的含钾化合物,这样的钾将最终残留在ADM 中。为此我们将初步探讨分析多膛焙烧钼精矿生产过程中产生和影响钾元素组成及其形态转变的因素,主要有以下几个方面:
1.1 反应温度
由于焙烧反应是强裂的放热反应。对于这个放热反应来说,温度升高易于造成焙烧熔融结块夹生,即产生高硫废品。
1.2 反应气氛
由于不断向炉内输入空气,气体始终是流动的,且二氧化硫的分压不会太高,那么氧化焙烧反应始终能进行完全,钼精矿中的硫能比较容易地脱除。
1.3 粒度对反应的影响
钼精矿颗粒愈小,且与氧接触条件愈好,反应速度愈快。
1.4 MoS2与MoO3的交互反应
在焙烧过程中,如果钼精矿烧结成块,则原来的气-固相反应的良好条件被恶化。虽然体系内仍保留有过剩的空气,但氧必须通过烧结块壳向其内部尚未氧化的MoS2界面扩散,因此供氧不足,在界面处MoS2和MoO3反应生成MoO2。
在钼精矿烧结块内部必定有一层MoO2,在MoO2层内就有可能存在尚未氧化的MoS2。可见,在焙烧过程中,应尽量避免钼精矿烧结成块,以保持气-固相反应的良好条件,确保焙烧去硫的效果。
1.5 共生硫化矿物的氧化
在相同的氧化焙烧条件下,钼精矿中硫化物的氧化顺序为FeS、MoS2、PbS、CuS、SnS。它的氧化物生成硫酸盐。钼精矿中硫化物杂质在焙烧中若生成硫酸盐时,因其分解温度高于焙烧温度,故不能分解,最后残留在钼精矿中,使之含硫量增加。因此,对钼精矿中硫化物杂质含量必须加以限制,特别是硫化铜、硫化铅能使钼精矿烧结,影响焙烧效果,必须严格限制[3]。
1.6 MoO3与杂质氧化物及钼精矿的反应
辉钼矿焙烧产生的MoO3可能与钼精矿中的杂质氧化物反应生成钼酸盐。CuMoO4和MoO3在560 ℃生成低熔点共晶,从而使钼精矿在焙烧过程中烧结,影响去硫效果[4]。
焙烧过程产生的CaMoO4和PbMoO4,因结块坚硬难溶于氨水,严重影响氨浸时钼的浸出率。
钼精矿中含有钾盐矿物(钾长石、杂卤石、钾石膏),由于熔融温度范围较大,熔体高温粘度较大,易于被钼精矿焙烧时产生的三氧化钼包裹粘接成块,这种包裹结块坚硬,其中含量极微的钾硫酸盐(杂卤石K2Ca2Mg(SO4)4·2H2O,钾石膏K2Ca2(SO4)2·H2O 是在SO2气氛中形成,因此无论对焙烧钼精矿的水洗还是氨浸作业,都将会对钼化工水洗或氨浸工艺造成严重影响,不但造成钼浸出率、回收率下降,也会使钼酸铵中钾铁等杂质含量超标,以至影响到后续钼金属生产产品质量。
1.7 适宜的负压
MoS2氧化反应强烈放热,而且反应本身放出的热量足以维持焙烧反应正常进行,所以就MoS2氧化焙烧成MoO3反应本身来说,不需外部供热。另外,MoO3熔点较低(795 ℃),若焙烧温度高于MoO3熔点,可见保证精矿不烧结也要求有过剩空气,使其带走部分反应热,降低反应温度。焙烧温度过高,变成气体随废气从炉内排出,造成钼金属的损失。因此,必须注意控制焙烧温度,同时还要回收废气中的MoO3。
2 多膛炉焙烧钼精矿工艺控制转变钾元素形态的主要途径
2.1 控制多膛炉收尾的顺序
焙烧炉中必须对2MoO2+O2=2MoO3,MoS2+6MoO3=7MoO2+2SO2两个反应进行控制使其完成。当反应将要完成时就进入了所谓的“收尾”时期,因此控制两个反应以正确的顺序完成将变得很关键。
根据不同的产品品质需要,可以通过观察9、10或11 号膛内的物料颜色来判断正确的收尾顺序,如图1 所示。如果控制适当,MoS2生成MoO2的反应较MoO2至MoO3的氧化反应较先完成,焙烧操作工通过观察旋转耙臂形成的料脊来判断出收尾顺序是否正确。如果收尾正确,将会观察到膛内物料出现灼热亮点,赤红色料脊,而后物料会变暗并松散沙化。
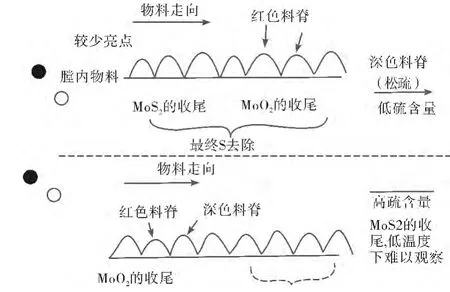
图1 多膛炉焙烧收尾示意图
2.2 控制或减少钼焙砂中Mo4O11及MoO2的产生
Mo4O11通常形成于9 号炉膛上下,Mo4O11在空气温度580 ℃左右开始熔化。熔化将形成粘性物质,粘在耙齿或在料膛中结球,结果会堵塞耙齿使得耙臂拖动整膛物料移动(即拖拽现象)。通过敞开清洁门试图解决“拖拽现象”只会适得其反,由于空气会进一步促进反应进而使物料温度更高,粘黏现象更为严重,发生拖拽现象的炉膛上方炉膛可能也会产生拖拽现象,因此控制9 号炉膛的温度极为关键。
2.3 应用产品冷却机并控制温度转变钼焙砂中钾的组织形态
有关资料表明:在指定炉膛温度下对氧化物保持较长时间对应ADM 产品中钾的浓度(以mg/kg表示)。如图2 所示水不溶NH4OH 溶解的含钾化合物将在525~600 ℃下形成。
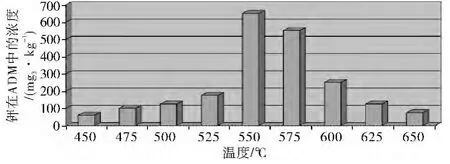
图2 多膛炉钼焙砂中钾含量与焙烧温度关系图
从图2 可以看出:多膛炉10~12 炉膛的缓慢冷却(630~450 ℃)会导致水不溶氨溶的含钾化合物增加。产品冷却器中将温度从12 炉膛的620 ℃快速降低100 ℃使产品很快通过危险区间(525~600℃)会防止这种化合物的生成。如此,产品将没有机会形成水不溶但氨溶的含钾化合物。急冷处理可以减少上述一种水不溶但NH4OH 可溶的含钾化合物产生,也就是说焙烧过程产生含量极微的钾硫酸盐在急冷条件下结晶组织状态发生变化且分散、颗粒疏松易于氨浸。
3 多膛炉实际焙烧生产过程钾元素变化检测分析
3.1 多膛炉实际焙烧生产过程钾元素变化检测分析结果
(1)试验与采样炉号及采样点位:1#多膛炉;1#多膛炉的12 层及产品冷却机出料口
(2)分析方法:钼精矿中钾分析用X 荧光光度分析法;1#多膛炉的12 层及产品冷却机出料口所采钼焙砂磨细制样后用质谱分析仪。金钼股份公司多膛炉实际焙烧生产过程钾元素变化检测分析结果见表1。
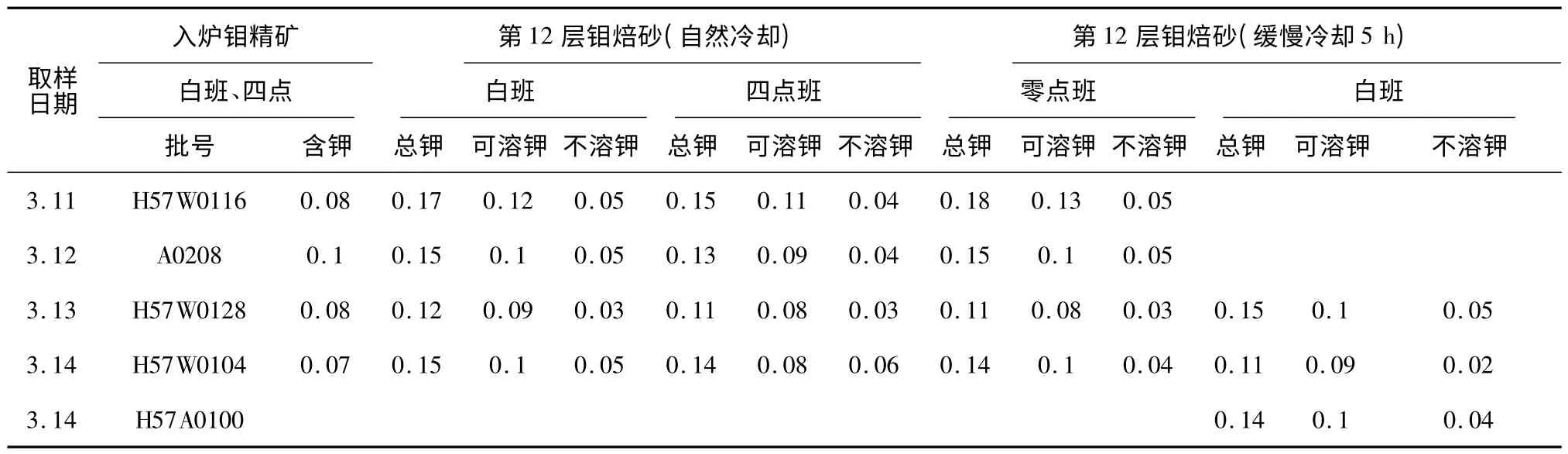
表1 多膛炉12 层钾元素变化检测分析结果 %
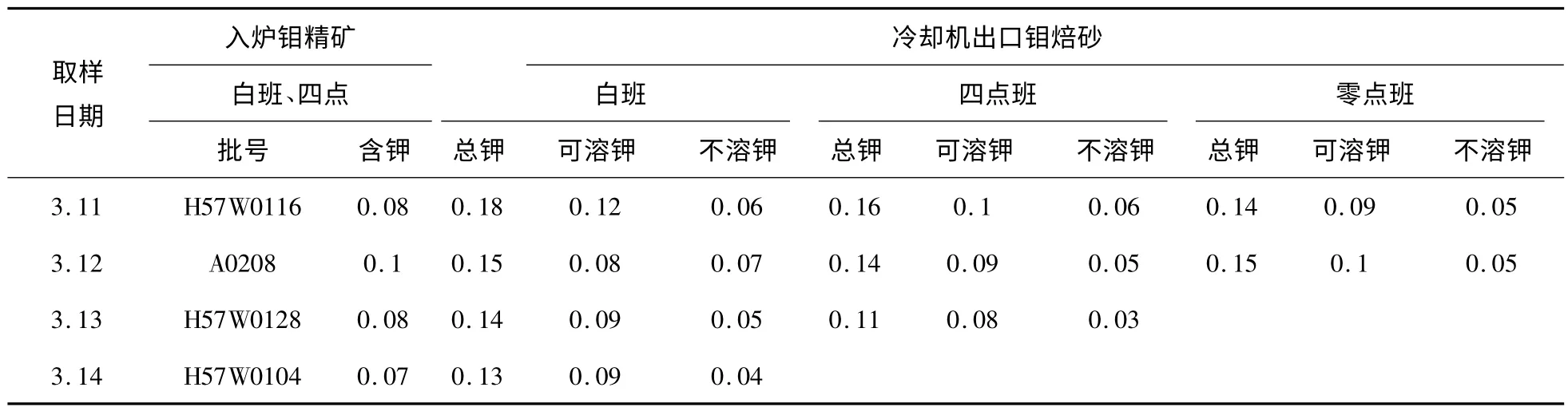
表2 多膛炉冷却机出口钾元素变化检测分析结果 %
3.2 分析结果讨论
多膛炉给料系统为2 个料仓,为1 开1 备,由于试验时多膛炉所存精矿料批号与炉台焙烧的批号始终不能一一对应,我们大致根据料仓内向多膛炉供给的批号与精矿在炉内停留反应时间推算加以对应,因而经采样分析的结果与实际有一定差距,因而试验结果讨论如下:
(1)是将钼精矿预处理的白班烘干批号采样而得,入炉钼精矿的含钾低于0.1%,最小值为0.07%,符合入炉钾含量要求。
(2)对多膛炉第12 层连续采样4 天,并采不同班次得到出炉焙砂自然冷却中含钾最大值0.18%,含钾最小值为0.11%,即入炉精矿焙烧后钾富集造成钾含量升高,但高于钼化工钾的含量要求值,可能有分析误差原因,也可有焙烧方面的原因。
(3)对多膛炉第12 层连续采样4 天,并采不同班次得到出炉焙砂将分别及时装入一耐火砖做成的保温箱保温5 h 取出样品装袋进行制样分析,得到的钼焙砂中含钾量最大值0.15%,最小值0.11%,与自然冷却值相差不大,但在钼化工钾的含量要求值的范围内。
(4)对多膛炉系统的产品冷却机出口连续采样4 天,并采不同班次得到出炉焙砂进行分析含钾最大值0.18%,含钾最小值0.11%,其这种情况与多膛炉第12 层连续采样4 天的结果几乎相同。
(5)反射炉与多膛炉焙烧高溶产品中钾含量相差变化不大,反射炉与多膛炉焙烧高溶产品二氧化钼含量同样无差别,但反射炉铜及铅含量较高,因此反射炉中含硫较多膛炉的高。
从上述情况可以得到,多膛炉第12 层与产品冷却剂含钾量几乎无变化,钾含量的变化与形态转变无任何关联,并且从焙烧钼精矿钾含量到钼精矿钾含量的富集系数为2.57,即钼精矿经焙烧后钾含量是上升的,因此焙烧过程对降钾的效果不明显,仅能减少焙烧结块以便提高后续氨浸时钼的浸出率。
4 结论及建议
(1)在加强控制多膛炉的温度和负压等参数的同时,必须控制多膛炉收尾的顺序以及减少M4O11的产生,提高钼精矿转变成高含量的三氧化钼,同时避免物料烧结结块产生包裹现象。
(2)控制多膛炉第12 层温度630~650 ℃,同时控制产品冷却机从第12 层炉膛的620 ℃快速降低100 ℃,控制冷却机循环水冷却水的温度,并使钼焙砂在产品冷却机中停留时间控制在5~7 min,这样可减少水不溶但NH4OH 可溶的含钾化合物产生。
(3)根据焙烧钼精矿原料进行XRD 分析发现含钾物质为云母、钾长石以及含量极微的钾硫酸盐,钾的来源为钾长石以及含量极微的钾硫酸盐,因此入炉钼精矿中的钾含量必须予以严格控制,进而减少对焙烧过程的危害和影响,乃至对钼化工系统工序的影响和危害。
(4)继续强化钼矿、焙烧钼精矿物相的测定与分析研究,为钼产业链各流程降钾提供强大的技术支持。
[1]赵新瑞,张增祥.工业钼粉中钾含量控制浅析[J].中国钼业,2010,34(4):44-46.
[2]任宝江.低钾钼粉的制备优化研究[J].中国钼业,2011,35(3):36-39.
[3]向铁根.钼冶金(修订版)[M].长沙:中南大学出版社,2002.
[4]李洪桂.稀有金属冶金学[M].北京:冶金工业出版社,1990.
