颈痹合剂的质量标准研究
2015-03-09魏晓舒张敏娟江苏省无锡药品检验所江苏无锡1408无锡市中医医院药剂科江苏无锡14071
魏晓舒,刘 燕,张敏娟(1.江苏省无锡药品检验所,江苏无锡 1408;.无锡市中医医院药剂科,江苏无锡 14071)
颈痹合剂是无锡市中医医院自制制剂,由羌活、葛根、木香等多味中药加工而成。该处方是成形的经验方,具有祛风除湿活血通络的功效,适用于风寒湿痹为主的颈型和神经根型颈椎病的治疗[1-2]。根据难治性疾病中药制剂的研发思路[3],并且根据《医疗机构制剂配制监督管理办法(试行)》[4]和《医疗机构制剂注册管理办法》[5]要求,本着确保特色制剂质量、提升医院制剂标准的宗旨,笔者采用薄层色谱(TLC)法对紫花前胡苷、去氢木香内酯、葛根素进行定性鉴别,采用高效液相色谱(HPLC)法对葛根的主要有效成分葛根素进行含量测定,并分析了制剂的相对密度、pH,为其质量控制提供一定参考。
1 材料
1.1 仪器
1200 型HPLC 仪,包括紫外检测器(美国Agilent 公司);AG-285 型电子天平、XP-6 型电子天平、MP220 型pH 计(瑞士Mettler-Toledo公司);ZF-20D型暗箱式紫外分析仪(上海顾村电光仪器厂)。
1.2 药品与试剂
颈痹合剂(无锡市中医医院自制,批号:110302、110927、110928、120104、120105、120106,规格:500 ml/瓶);葛根素对照品(批号:110752-200912,纯度:96%)、去氢木香内酯对照品(批号:111525-200505,纯度>98%);紫花前胡苷对照品(批号:111821-201102,纯度>98%)均购自中国食品药品检定研究院;硅胶G薄层板(青岛海洋化工厂)乙腈为色谱纯,其余试剂均为分析纯,水为超纯水。
2 方法与结果
2.1 定性鉴别
2.1.1 羌活 取样品20 ml,加乙醚振摇提取2 次(每次20 ml),合并乙醚液备用;水液加水饱和正丁醇振摇提取2 次(每次20 ml),合并正丁醇液,蒸干,残渣加甲醇2 ml使溶解,作为供试品溶液。取紫花前胡苷对照品适量,加甲醇溶解制成每1 ml含0.5 mg的对照品溶液。按处方及制备工艺制备缺羌活的阴性对照样品,按上述供试品溶液制备方法制成阴性对照溶液。按上述TLC 法[2010 年版《中国药典》(一部)附录ⅥB]试验,吸取上述3 种溶液各10 μl,分别点于同一硅胶G 薄层板上,以三氯甲烷-甲醇(4∶1,V/V)为展开剂,置氨蒸气饱和的展开缸内,展开,取出,晾干,置紫外光灯(365 nm)下检视。结果显示,供试品色谱中,在与对照品色谱相应位置上显相同颜色的斑点,且阴性对照无干扰,详见图1A。
2.1.2 木香 取“2.1.1”项下乙醚液,低温挥去乙醚,残渣加三氯甲烷2 ml 使溶解,作为供试品溶液。取去氢木香内酯对照品适量,加三氯甲烷溶解制成每1 ml 含0.5 mg 的对照品溶液。按处方及制备工艺制备缺木香的阴性对照样品,按供试品溶液制备方法制成阴性对照溶液。按TLC法[2010年版《中国药典》(一部)附录ⅥB]试验,吸取上述3种溶液各10 μl,分别点于同一硅胶G薄层板上,以环已烷-甲苯-乙酸乙酯(2∶1∶1,V/V/V)为展开剂,置氨蒸气饱和的展开缸内,展开,取出,晾干,喷以1%香草醛硫酸溶液,热风吹至斑点显色清晰,置日光下检视。结果显示,供试品色谱中,在与对照品色谱相应位置上显相同颜色的斑点,且阴性对照无干扰,详见图1B。
2.1.3 葛根 取样品30 ml,用氢氧化钠调pH 至9~10,加乙酸乙酯振摇提取2次(每次20 ml),弃去乙酸乙酯液;水液加稀盐酸调pH 至5~6,用乙酸乙酯振摇提取2 次(每次30 ml),合并提取液,经无水硫酸钠2 g滤过,滤液蒸干,残渣加甲醇2 ml使溶解,作为供试品溶液。按处方及制备工艺制备缺葛根的阴性对照样品,按供试品溶液制备方法制成阴性对照溶液。取葛根素对照品适量,加甲醇制成每1 ml 含0.5 mg 的对照品溶液。吸取上述溶液各10 μl,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水(7∶2.5∶0.25,V/V/V)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。结果显示,供试品色谱中,在与对照品色谱相应的位置上显相同颜色的荧光斑点,且阴性对照无干扰,详见图1C。
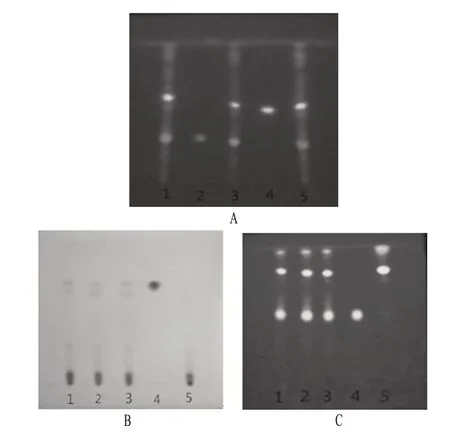
图1 薄层色谱图A.羌活[1、3、5.供试品(批号:120104、120105、120106);2.阴性对照;4.对照品];B.木香[1~3.供试品(批号:120104、120105、120106);4.对照品;5.阴性对照];C.葛根[1~3.供试品(批号:120104、120105、120106);4.对照品;5.阴性对照]Fig 1 TLC chromatogramsA.Notopterygii Rhizoma et Radix[1.3.5.test samples(batch number:120104,120105,120106);2.negative control;4.reference substance];B.Aucklandia lappa[1-3.test samples(batch number:120104,120105,120106);4.negative control;5.reference substance];C.Pueraria lobata[1-3.test samples(batch number:120104,120105,120106);4.negative control;5.reference substance]
2.2 相对密度和pH测定
按2010年版《中国药典》(一部)附录ⅧA 相对密度测定法比重瓶法测定样品的相对密度。结果显示,6批样品的相对密度平均值为1.08,RSD 为0.6%(n=6)。按2010 年版《中国药典》(一部)附录ⅧG pH 测定法测定样品pH。结果显示,6 批样品pH平均值为4.5,RSD为1.5%(n=6)。
2.3 含量测定
2.3.1 色谱条件与系统适用性试验 色谱柱:XTer-ra®RP18(250 mm×4.6 mm,5 μm);流动相:乙腈-0.05%磷酸(10∶90,V/V);流速:1.0 ml/min;检测波长:250 nm;柱温:25 ℃;进样量:10µl。当信噪比为12∶1、3.9∶1时,葛根素定量限、检测限分别为0.33、0.17 μg/ml。在上述色谱条件下,理论板数以葛根素峰计应不低于5 000,分离度应不小于1.5,详见图2。
2.3.2 对照品溶液的制备 分别精密称取葛根素对照品1.257、1.732 mg,用30%乙醇分别制成每1 ml 含葛根素0.120 7、0.166 3 mg 的对照品贮备液Ⅷ、Ⅷ。精密吸取对照品贮备液Ⅷ适量,用30%乙醇制成每1 ml 含葛根素6.034 μg 的对照品溶液。
2.3.3 供试品溶液的制备 精密吸取样品5 ml,置于100 ml量瓶中,用30%乙醇定容,摇匀,静置1 h,取上清液,滤过;精密吸取续滤液1 ml,置于10 ml 量瓶中,用30%乙醇定容,摇匀,滤过,取续滤液,即得。

图2 高效液相色谱图A.阴性对照;B.对照品;C.供试品;1.葛根素Fig 2 HPLC chromatogramsA.negative control;B.reference substance;C.test sample;1.puerarin
2.3.4 阴性对照溶液的制备 取不含葛根的其他药味各适量,按颈痹合剂的制备工艺制成缺葛根的阴性样品,再按“2.3.3”项下方法制成阴性对照溶液。
2.3.5 线性关系考察 分别精密量取“2.3.2”项下葛根素对照品贮备液Ⅷ适量,用30%乙醇制成质量浓度分别为33.26、16.63、8.315、4.158、2.079 μg/ml的系列对照品溶液。精密吸取上述系列对照品溶液各10 μl,按“2.3.1”项下色谱条件进样6次测定,记录峰面积。以葛根素质量浓度(x,μg/ml)为横坐标、峰面积(y)为纵坐标进行线性回归,得回归方程为y=44.048x-14.042(r=0.999 8,n=6)。结果表明,葛根素检测质量浓度线性范围为2.079~33.26 μg/ml。
2.3.6 精密度试验 取“2.3.2”项下对照品溶液适量,按“2.3.1”项下色谱条件连续进样测定6 次,记录峰面积。结果,葛根素峰面积的RSD为0.14%(n=6),表明仪器精密度良好。
2.3.7 稳定性试验 取“2.3.3”项下供试品(批号:110302)溶液适量,分别于放置0、3、6、9、12 h 时进样测定,记录峰面积。结果,葛根素峰面积的RSD 为0.2%(n=5),表明供试品溶液在12 h内基本稳定。
2.3.8 重复性试验 精密称取同一批样品(批号:110302)适量,按“2.3.3”项下方法制备供试品溶液,共6份,再按“2.3.1”项下色谱条件进样测定,记录峰面积。结果,葛根素峰面积的RSD为0.2%(n=6),表明本方法重复性良好。
2.3.9 加样回收率试验 取已含量的样品(批号:110302)适量,共6份,分别加入一定量葛根素对照品,按“2.3.3”项下方法制备供试品溶液,再按“2.3.1”项下色谱条件进样测定,计算样品含量,并计算加样回收率,结果见表1。
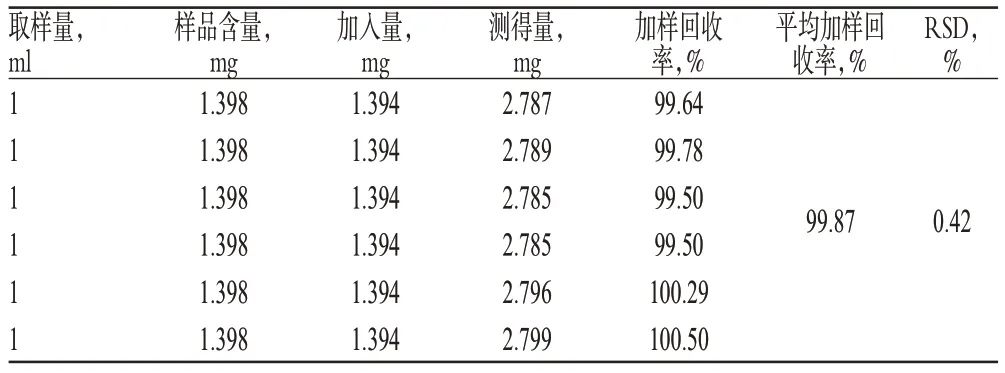
表1 加样回收率试验结果(n=6)Tab 1 Results of recovery test(n=6)
2.3.10 样品含量测定 取6 批样品各适量,分别按“2.3.3”项下方法制备供试品溶液,再按“2.3.1”项下色谱条件进样测定,计算样品含量,结果见表2。

表2 样品含量测定结果(n=3)Tab 2 Results of content determination of samples(n=3)
3 讨论
颈痹合剂属多组分制剂,其中主药羌活中紫花前胡苷、木香中去氢木香内酯和葛根中葛根素均具有鉴别的特征性意义,而处方中其他药材均不含这些特征成分[6],因此选择该三种成分进行TLC 鉴别。经过对供试品溶液前处理、展开剂的选择及显色方式筛选,该色谱结果专属性较强、斑点清晰、阴性对照无干扰,这对原药材的质量及制备工艺都起到了监督作用。
有研究报道,HPLC测定颈痹颗粒中葛根素含量时流动相采用甲醇-水(25∶75,V/V)[7]。本研究的预试验结果表明,针对以水为溶剂的供试品溶液,应采用乙腈-水配比的流动相,用适量磷酸调节可使葛根素峰形对称,清除杂峰能力增强,重现性较好。当水比例占很大部分时,不宜采用一般的ODS色谱柱,而利用屏蔽技术的XTerra®RP色谱柱有很好的水浸润性,即便是使用100%的水溶液作为流动相,亦可获得稳定的色谱结果。2010 年版《中国药典》(一部)规定:葛根药材按干燥品计算,葛根素含量不得少于2.4%[8],这为制定颈痹合剂中葛根素定量指标提供了参考依据,也给制备工艺提出了新要求。
综上所述,该方法操作简便、重复性好,可用于颈痹合剂的质量控制。
[1]张兴国,周文浩.颈痹合剂治疗颈型及神经根型颈椎病80 例临床观察[J].长春中医药大学学报,2011,27(2):263.
[2]吴震海.颈痹合剂治疗颈型及神经根型颈椎病的疗效观察[J].临床合理用药,2012,5(3A):79.
[3]宋洪涛,张晶,周欣,等.当前医院制剂发展策略与研发思路探讨[J].中国药房,2009,20(13):999.
[4]国家食品药品监督管理局.医疗机构制剂配制监督管理办法:试行[S].2005-06-01.
[5]国家食品药品监督管理局.医疗机构制剂注册管理办法:试行[S].2005-08-01.
[6]肖培根.新编中药志:第一卷[M].北京:化学工业出版社,2002:121.
[7]何建华,钱永昌,许勇.HPLC 测定痉痹颗粒中葛根素的含量[J].海峡药学,2005,17(6):81.
[8]国家药典委员会.中华人民共和国药典:一部[S].2010年版.北京:中国医药科技出版社,2010:313.
