运动性细胞自噬是调节骨骼肌代谢稳态的内置机制
2015-02-14钱帅伟丁树哲
钱帅伟,丁树哲
运动性细胞自噬是调节骨骼肌代谢稳态的内置机制
钱帅伟1,2,丁树哲1
细胞自噬作为骨骼肌必需的代偿性内置调节机制,可在运动、禁食、营养限制和肌肉收缩刺激等能量应激下,将胞浆中损伤或衰老的细胞组件(线粒体、内质网、核糖体)、病菌和ROS等代谢废物,以及糖原、脂质、非功能或功能性蛋白质等能源物质,转运到溶酶体中消化降解,从而完善骨骼肌细胞质量控制,有效供给细胞更新和代谢平衡所需的能量与合成底物的一种分解代谢装置。运动训练不仅能通过细胞自噬完善骨骼肌线粒体质量控制,稳定线粒体功能网络,维持骨骼肌代谢稳态,还能有效防治胰岛素抵抗、肥胖和II型糖尿病等代谢疾病的发生。运动训练介导的细胞自噬也可使骨骼肌质量及其功能根据运动项目的自身特点进行积极调整和适应,从而进一步维持骨骼肌代谢功能稳态。
细胞自噬;骨骼肌;运动训练;代谢稳态;线粒体质量控制;代谢疾病;肌肉质量
骨骼肌具有高度可塑性,其形态、结构和功能可随着多种生理或病理性的持续刺激(如运动、运动不足、去神经支配、电刺激和低氧等)而产生适应性改变。运动作为一种经典的生理性刺激方式,可促发骨骼肌进行持续频繁收缩,使其形态结构和生理功能根据运动项目的本质特点进行积极调整,并产生整体适应和良性重塑。一般来说,耐力运动可使骨骼肌新生血管增多,肌糖原储量增加,氧化型肌纤维转化率提高,线粒体含量增加、功能改善,从而防治冠心病和高血压等心血管疾病[11]。抗阻运动可使肌纤维增粗,横截面积增大,肌肉质量和体积增加,从而防范衰老所致的骨骼肌流失、肌力下降和基础代谢率失调[11]。而同期进行耐力运动和抗阻运动则可有效防治肥胖、代谢综合征和II型糖尿病所致的胰岛素抵抗和肌肉功能异常[9]。因此,不同方式运动驯化的骨骼肌结构和功能的积极适应与重塑是维持骨骼肌健康乃至整个机体健康的重要前提与基础。
尽管运动可对骨骼肌乃至整个机体带来积极的健康效益,但骨骼肌收缩或运动也产生了许多负面效应,如代谢副产物ROS的产生、衰老或错误折叠蛋白质的累积、非功能或损伤细胞组件(线粒体、核糖体和内质网等)的聚集等[54]。这些代谢废物若得不到及时清除和有效降解,不仅会减损运动带来的积极健康效益,还可能导致骨骼肌代谢功能紊乱,甚至诱发细胞凋亡或死亡,从而损害骨骼肌健康。这说明,骨骼肌迫切需要通过一种内源性代偿调节装置来清除这些代谢废物,甚至产生能量底物,从而有效维持肌肉收缩和能量代谢稳态。
近期研究充分表明,细胞自噬作为骨骼肌细胞中普遍存在的代谢现象,可在禁食、营养限制、运动和肌肉收缩刺激等能量应激下,将胞浆中损伤或衰老的细胞器、病菌和ROS等代谢废物,以及非功能或功能性蛋白质、脂质和糖原等能源物质,转运到溶酶体中消化降解,从而完善骨骼肌细胞质量控制,提供细胞更新和代谢平衡所需能量与合成底物的一种代谢装置[45]。运动训练不仅能通过细胞自噬完善骨骼肌线粒体质量控制,稳定线粒体功能网络,维持骨骼肌代谢稳态,还能有效防治胰岛素抵抗、肥胖和II型糖尿病等代谢疾病发生。运动训练也可使骨骼肌质量及其功能根据运动项目的自身特点进行积极调整和适应,从而进一步稳定骨骼肌代谢功能稳态。因此,自噬在骨骼肌代谢功能稳态调控方面具有不可或缺的重要作用。
1 细胞自噬是调节骨骼肌细胞代谢稳态的内置机制
骨骼肌不仅是机体最主要的运动应答器官,也是物质能量代谢的重要场所。骨骼肌收缩时,其能量需求急剧递增,因此,能量的高效产出和稳定供给是维持骨骼肌代谢稳态的重要保证。营养充足或基础状态时,葡萄糖主要以糖原形式储存在肝脏或骨骼肌中。葡萄糖饥饿或中、高等强度运动时,糖原可在糖原磷酸化酶作用下,水解产生游离葡萄糖,供肌细胞摄取和利用。但这种经典过程并非游离葡萄糖产生和释放的唯一机制。自噬在糖原降解过程中也同样扮演不可或缺的重要角色。耐力运动可增强胰高血糖素的分泌能力,后者作为糖原降解的重要调节激素,可增强溶酶体酸性糖苷酶的活性,促使糖原通过自噬途径降解,并产生游离葡萄糖,供骨骼肌收缩需要[27]。糖原自噬障碍可致自噬体或溶酶体中糖原异常储积,诱发肌病(Pompe和Danon病),而重新激活自噬则可有效缓解糖原负荷[46]。He等[19]研究发现,急性和耐力运动均可使野生型小鼠骨骼肌细胞自噬水平上升,糖代谢能力增强;但通过建立Bcl-2AAA(Thr69/Ser70/Ser84磷酸化位点缺失)自噬缺陷小鼠模型,发现急性运动或营养缺乏由于不能上调Bcl-2AAA小鼠骨骼肌自噬水平,致使葡萄糖转运体4(glucose transporter type 4,GLUT4)转运能力降低,葡萄糖耐受力下降,糖代谢平衡紊乱,运动耐力水平降低。提示,自噬对于维持骨骼肌糖代谢稳态具有重要作用。肌细胞甘油三酯(intramyocellular triacylglycerol,IMTG)仅占机体总储脂的1%~2%,但在90 min的中等强度运动时,可提供高达25%的能量供应[11]。能量匮乏和耐力运动均可使IMTG在脂肪甘油三酯酶(adipose triglyceride lipase,ATGL)和激素敏感性脂肪酶(hormone sensitive lipase,HSL)作用下,分解产生自由脂肪酸,供肌细胞氧化利用[27]。自噬也是促进自由脂肪酸产生和释放的重要代谢装置。禁食、饥饿或运动时,骨骼肌可通过脂质自噬,将脂滴裹入自噬体,随后被溶酶体酸性脂肪酶降解为游离脂肪酸,供肌肉收缩利用。自噬功能异常可使IMTG降解障碍,脂滴在胞浆过度储积,降低脂肪酸β氧化和ATP产出率,导致骨骼肌代谢紊乱,甚至诱发血脂异常、肥胖和II型糖尿病等代谢疾病发生。尽管葡萄糖和自由脂肪酸是骨骼肌收缩时的重要供能物质,蛋白质或氨基酸也是必不可少的能量底物。中等强度运动时,蛋白质代谢的能量供应约占5%~15%,甚至90 min的高强度运动时,其可提供高达20%的能量供应[11]。骨骼肌作为机体最大的蛋白质储存库,可在能量耗竭或长时间运动时,通过自噬途径降解肌肉蛋白质,产生氨基酸等能量底物,为肌肉收缩提供能量来源,或供合成新的蛋白质。能量匮乏时,骨骼肌、肝脏等组织可通过自噬途径降解蛋白质,产生谷氨酰胺、丙氨酸等多种氨基酸,释放入血液,为自身或其他组织提供充足的能量来源[27]。这说明,自噬介导的蛋白质水解对于维持氨基酸代谢水平和能量稳态具有至关重要的作用。此外,自噬也可通过降解胞浆中损伤的细胞器、ROS等代谢废物,从而有效维持骨骼肌质量和力量。骨骼肌Col6a1-/-小鼠由于自噬缺陷而使肌浆网扩张,空泡化,损伤细胞器异常聚集,氧化应激水平提高,并出现肌肉萎缩和肌力下降等症状,即使通过递增跑台训练也不能逆转自噬水平的降低,以及肌肉质量和力量的下降[17]。
这说明,细胞自噬既可降解胞浆中储积的能源物质,产生多种能量底物,为肌肉收缩或代谢提供稳定的能量来源;也可回收胞浆中聚集的代谢废物,从而进一步维持骨骼肌质量,稳定骨骼肌代谢功能稳态。自噬通路障碍可引起整个机体代谢功能紊乱,导致肌肉衰减征、胰岛素抵抗、肥胖、II型糖尿病和衰老等代谢疾病发生。因此,细胞自噬是骨骼肌代谢稳态必需的内置调节机制。
2 骨骼肌细胞自噬关键激活通路
2.1 mTOR/ULK1信号轴
禁食、能量匮乏、去神经支配、低氧和运动等外源性应激刺激均可诱导细胞自噬[13]。最经典的自噬诱导途径是通过哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)与ULK1(UNC51-like kinase)复合物之间的相互作用来实现的。mTOR是一种保守的丝/苏氨酸蛋白激酶,在调节细胞生长、分化、增殖、凋亡和自噬等方面具有重要作用。ULK1复合物主要由ULK1、FIP200和Atg13等自噬蛋白组成,该复合物的形成是诱导自噬的前提与关键[60]。正常生理情况下,mTOR可磷酸化抑制ULK1和Atg13的活性,阻止ULK1复合物形成,使自噬保持基础水平;但生长因子下降、营养匮乏时,mTOR受到抑制,从而解除其对ULK1和Atg13的磷酸化,形成ULK1-Atg13-FIP200复合物,诱导细胞自噬[25]。
氨基酸、生长因子等应激性刺激可选择性激活磷脂酰肌醇3-激酶(phosphotidylinositol 3-kinase,PI3K)/蛋白激酶B(protein kinase B,Akt)通路,Akt被激活后,可磷酸化TSC1/2 Thr1462位点,并通过与Rheb-GTP作用,激活mTOR,后者可磷酸化ULK1 Ser757位点,抑制其与Atg13的结合能力,从而抑制ULK1-Atg13-FIP200复合物形成,减弱自噬活性[25]。腺苷酸活化蛋白激酶(AMP activated protein kinase,AMPK)作为能量状态的敏感感受器,是细胞自噬的重要激活因子。缺血、低氧、营养匮乏、禁食、急性或耐力运动均可引起骨骼肌细胞环境急剧变化,使AMP/ATP比率升高,进而激活AMPK,后者可磷酸化Raptor Ser722/Ser792位点,募集14-3-3蛋白与Raptor结合,从而抑制mTOR,解除其对ULK1的磷酸化,激活细胞自噬;或通过磷酸化TSC2 Thr1227/Ser1345位点,进而抑制mTOR,诱导细胞自噬[13,54]。AMPK也可直接磷酸化ULK1 Ser317/Ser555/Ser467/Ser637/Ser777位点,增强ULK1的活性,促进ULK1-Atg13-FIP200复合物形成,诱导细胞自噬[54]。
但近期研究发现,mTOR也可通过ULK1非依赖性途径调节自噬水平。转录因子EB(transcription factor EB,TFEB)在溶酶体生物发生和自噬激活等方面具有重要作用[57]。基础状态下,V-ATPase、Rag GTPase等因子可募集mTOR至溶酶体膜,并磷酸化TFEB Ser142/Ser211位点,将TFEB阻滞在胞浆,从而抑制溶酶体生物发生;但能量匮乏可抑制mTOR,使其从溶酶体上解离,重新恢复TFEB的核定位,促进溶酶体生物发生和自噬激活[57](图1)。
2.2 Bcl-2/Beclin1复合体
Bcl-2/Beclin1复合体是细胞自噬的另一条重要激活通路。Beclin1作为酵母自噬基因Atg6/Vps30的同源物,可通过其自身结构域与Vps34、Vps15、UVRAG和Ambra1等蛋白相互作用,并组成复合体,促进自噬体形成与成熟,增强自噬活性,但该通路却受到抗凋亡蛋白Bcl-2的调控[52]。
正常生理情况下,Bcl-2与Beclin1结合能力较强,从而抑制Beclin1与Vps34、Vps15等自噬蛋白形成复合体,使自噬保持基础水平。但氧化应激、营养匮乏等外源性刺激可引起骨骼肌c-JunN末端激酶JNK1的激活,并磷酸化Bcl-2,导致Bcl-2/Beclin1复合物的有效解离与Beclin1的释放,从而激活细胞自噬[19,23]。He等[19]通过建立Bcl-2AAA自噬缺陷小鼠模型,发现饥饿、急性或耐力运动由于不能磷酸化骨骼肌和心肌Bcl-2,使Beclin1与Bcl-2不能成功解离,自噬活性严重削弱,糖代谢平衡紊乱,运动耐力水平降低。McMillan等[43]研究也发现,耐力性有氧运动可磷酸化骨骼肌和心肌Bcl-2 Ser87位点,降低Bcl-2蛋白含量,提高p-Bcl-2/Bcl-2比率,促进Bcl-2/Beclin1复合体的解离和Beclin1的释放,激活细胞自噬,且认为这是独立于AMPK/ULK1、Akt/mTOR/ULK1和Akt/FoxO3等通路的新型自噬诱导途径。AMPK也是Bcl-2/Beclin1复合物的重要调节因子。葡萄糖饥饿时,AMPK可磷酸化Vps34 Thr163/Ser165位点,抑制胞浆中不参与自噬的Vps34复合物,并磷酸化Beclin1 Ser91/Ser94位点,促进参与自噬的Vps34复合物的形成,上调细胞自噬[24]。
但近期研究发现,氨基酸匮乏或mTOR受到抑制时,ULK1可与UVRAG结合,并磷酸化Beclin1 Ser14位点,促进Bcl-2与Beclin1的解离,形成Beclin1-Atg14L-Vps34复合物,诱导细胞自噬[52]。这说明,Bcl-2/Beclin1复合体也可通过mTOR/ULK1依赖性方式,诱导细胞自噬(图1)。

图1 本研究mTOR/ULK1和Bcl-2/Beclin1调控骨骼肌细胞自噬的分子机制示意图
2.3 FoxO3
叉头框蛋白O3(forkhead box protein O3,FoxO3)属于Forkhead蛋白家族的重要成员之一,在细胞增殖、分化、凋亡和自噬等方面具有重要作用。以前研究表明,FoxO3主要通过转录激活肌萎缩相关基因atrogin-1和MuRF1的表达,通过泛素-蛋白酶体途径降解肌肉蛋白质[45,56]。但近期研究发现,耐力运动、去神经支配和禁食时,FoxO3也可转录激活自噬相关基因LC3、Gabarapl1、Bnip3、ULK2、Atg4、Atg5、Atg12和Atg16的表达,诱导细胞自噬[21,40]。这说明,FoxO3诱导的骨骼肌细胞自噬也是独立于mTOR/ULK1的重要激活通路。
FoxO3的翻译后修饰也是调节骨骼肌自噬通量水平的重要因素[60]。能量充裕时,Akt可磷酸化FoxO3 Thr32/Ser315/Ser253位点,阻遏其入核,抑制其介导的自噬基因转录,从而下调自噬水平[55]。Reynolds等[49]研究表明,Akt1/2-/-小鼠骨骼肌Bnip3和Gabarapl1表达升高,提示,Akt是FoxO3介导细胞自噬的重要抑制因子。AMPK在FoxO3介导的自噬通路中也具有重要作用。AMPK可磷酸化FoxO3 Ser588/Ser413位点,使其转录活性增强,促进细胞自噬[55]。沉默信息调节因子1(Sirtuin1,SIRT1)是一种烟酰胺腺嘌呤二核苷酸(NAD+)依赖的组蛋白去乙酰化酶。在胞浆中,SIRT1可直接去乙酰化激活Atg5、Atg7、Atg12和LC-3等自噬蛋白,诱导细胞自噬;但在胞核中,SIRT1则可去乙酰化多种转录因子,如p53、NF-kB、FoxO1、FoxO3和PGC-1α等,从而调节自噬水平[13]。禁食或运动可激活骨骼肌AMPK,并通过调节NAD+/NADH的比率,促进SIRT1依赖的FoxO3、FoxO1和PGC-1α去乙酰化激活,从而诱导细胞自噬,产生能量底物[7,13]。CBP/P300是一种组蛋白乙酰化酶,可介导细胞生长、分化、凋亡、自噬和DNA损伤修复等过程[29],其不仅能乙酰化LC-3、Atg5、Atg7和Atg12等自噬蛋白,也可乙酰化抑制FoxO3及其介导的自噬基因转录,从而下调自噬水平[29]。
miRNAs是近期研究较多的一类非编码小RNA分子,可通过降低其靶基因mRNA稳定性和翻译抑制的方式参与靶基因表达的调控。C2C12肌细胞转染miR-182可靶向抑制FoxO3 mRNA和蛋白表达,并下调atrogin-1、Atg12、Cathepsin L和LC-3的表达[20]。体内研究也证实,糖尿病大鼠骨骼肌miR-182水平的降低可使FoxO3过量表达,引起自噬过度激活,导致肌肉萎缩和肌力下降[20]。这说明,miRNAs也是FoxO3介导自噬通路的重要调节因子(图2)。
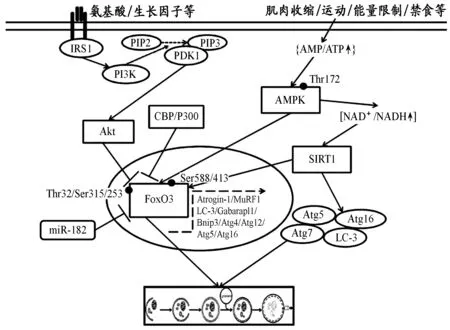
图2 本研究FoxO3调控骨骼肌细胞自噬的分子机制示意图
3 运动训练通过细胞自噬调节骨骼肌代谢功能稳态的内置机制
线粒体是一种双层膜封闭式细胞器,其形态、结构、数量和质量具有高度可塑性。线粒体也是生物氧化和能量交换的重要场所,可通过三羧酸循环和氧化磷酸化产生ATP,为生命活动提供能量和生物合成底物[11]。肌肉收缩或运动时,其能量需求急剧递增,这些能量主要来自线粒体氧化代谢。因此,促进线粒体生物发生或维持其质量控制是稳定线粒体功能,保证肌肉收缩所需能量供应的重要保证。
PGC-1α被公认为运动诱导线粒体生物发生的万能调节因子。急性运动[8]、耐力运动[59]和高强度间歇运动[33]均可促进骨骼肌PGC-1α mRNA和蛋白表达,后者可辅助雌激素相关受体γ(estrogen-related receptor γ,ERRγ)、核呼吸因子2( nuclear respiratory factor 2,NRF2)、NRF1和线粒体转录因子A(transcription factor A mitochondria,TFAM)等因子激活核编码或线粒体编码的基因,从而促进线粒体生物发生,增加线粒体数量和体积。运动通过PGC-1α诱导的线粒体生物发生也受到多种信号通路的正性调节,如p38 MAPK/ATF2、Ca2+/钙调磷酸酶(calcineurin,CaN)和Ca2+/CaMKIV,以及AMPK的磷酸化和SIRT1的去乙酰化修饰等[51]。
但运动通过促进线粒体生物发生供给能量的同时,也产生了副产物ROS。适量水平的ROS可作为信号分子参与细胞信号转导,调节细胞生长、分化和存活,甚至对外源性病原体也有杀伤或清除作用,但过量的ROS则可攻击DNA、蛋白质和脂质,损害线粒体结构和功能的完整性,使损伤线粒体异常累积,并产生更多ROS,导致过氧化连锁反应,甚至启动线粒体介导的凋亡[13]。因此,为了稳定细胞的正常活动状态,线粒体亟需通过融合与分裂循环进行组分重组,使受损或功能失调的线粒体隔离出线粒体网络,并进行特异性修复或清除,从而维持线粒体结构和功能的完整性。线粒体融合分裂事件主要涉及线粒体融合关键蛋白(Mfn1/2、Opa1等)和分裂关键蛋白(Drp1、Fis1等)的参与[2]。这些蛋白的协同配合共同维持线粒体结构的动力学变化。运动促发的能量代谢环境的剧烈变化可引起线粒体融合分裂的动态调整。耐力运动、高强度间歇运动均可促进骨骼肌Mfn1/2和Opa1的表达,提示,线粒体融合是线粒体对能量需求急剧变化的一种快速应答机制[2]。但也有研究表明,耐力运动可促进骨骼肌Mfn2 mRNA表达和Drp1的磷酸化[51],使线粒体融合与分裂能力均进行动态调整和适应。而后肢去负荷或运动不足则可抑制Mfn2和Drp1的表达[48],降低线粒体融合分裂能力。甚至Garnier等[15]研究认为,耐力训练可诱导骨骼肌Mfn2和Drp1的表达,这与PGC-1α升高呈正相关。但总体而言,耐力运动可改变线粒体融合分裂的动态平衡能力,但使其更倾向于融合,从而增加线粒体含量(代谢底物、线粒体DNA和蛋白质等),且这种变化很可能受PGC-1α驱动;肌肉废用或运动不足则使线粒体更趋向于分裂,使线粒体呈碎裂或断裂化,致使能量代谢紊乱,ATP产出降低,ROS产生增多[64]。
线粒体的动态变化分裂最终产生两个不均匀的子代,其中,膜电位严重衰减的线粒体子代,以及超出自身修复能力的线粒体,可通过自噬途径选择性降解(即线粒体自噬)。线粒体自噬是指在ROS、营养匮乏和细胞衰老等应激性刺激下,线粒体产生去极化损伤,随后被裹入自噬体,并与溶酶体融合,完成损伤线粒体消化降解,从而维持线粒体稳态的一种代谢装置[28]。目前普遍认为,Pink1、Parkin和Bnip3/Nix是参与线粒体修复及线粒体自噬的特异性蛋白。Pink1是一种丝/苏氨酸蛋白激酶,其在健康线粒体中可被线粒体蛋白水解酶迅速降解;但损伤或衰老线粒体Pink1降解机制障碍,致使Pink1过度累积,其可磷酸化并募集Parkin选择性转位到受损线粒体,而后介导Mfn1/2和Drp1等蛋白的泛素化,参与线粒体自噬[28]。Bnip3/Nix主要位于线粒体外膜,可与自噬蛋白LC-3相互作用,介导线粒体靶向转入自噬体消化降解。自噬对线粒体的选择性降解决定了自噬相关基因的表达具有肌纤维特异性,由于LC3B、Gabarapl1、Bnip3和Parkin是参与线粒体自噬的关键蛋白,故其主要分布于高氧化型骨骼肌(如比目鱼肌),而ULK1、Atg5和Atg12等自噬蛋白主要分布于糖酵解型骨骼肌(胫骨前肌、趾长伸肌和腓肠肌等)[54]。早期研究[53]发现,小鼠进行剧烈跑台训练后,胞浆自噬泡出现不同降解阶段的线粒体,提示,自噬可降解损伤线粒体等亚细胞组件,为肌纤维再生提供组装底物。Lira等[32]研究发现,耐力训练可显著增强骨骼肌Atg7、Beclin1和LC3-II等自噬蛋白的表达,提高LC3-II/LC3-I比率,降低P62/SQSTM1蛋白含量,并可促进线粒体自噬蛋白Bnip3表达,改善骨骼肌基础自噬和线粒体自噬水平,降解损伤或衰老线粒体,稳定线粒体代谢适应和运动耐力水平。崔迪等[1]研究发现,长期高脂膳食可降低骨骼肌Bnip3/Nix mRNA表达,但耐力训练可增强高脂膳食小鼠骨骼肌Pink1、Parkin和Nix/Bnip3的表达,诱导线粒体自噬,降解损伤或衰老线粒体,稳定线粒体功能。这说明,耐力运动诱导的线粒体自噬是促进线粒体进行更新循环和功能改善的重要内置机制。
但更多研究认为,运动诱导的线粒体更新循环和功能改善依赖于线粒体自噬与线粒体生物发生、融合分裂循环的耦联发生和协调适应。Guo等[18]研究发现,睾酮+低强度耐力训练既可促进线粒体融合分裂基因Mfn2和Drp1的表达,也可增强线粒体自噬蛋白LC3-II和Parkin的表达,从而稳定线粒体质量控制,抑制骨骼肌氧化损伤。Jamart等[22]研究表明,急性低强度耐力运动可增强禁食状态小鼠骨骼肌PGC-1α、LC3b-II、Atg12、Drp1、Bnip3和Parkin的表达,增加LC3b-II/LC3b-I比率,促进线粒体生物发生、融合分裂和线粒体自噬,稳定线粒体质量控制。Vainshtein等[63]研究表明,急性力竭运动可促进骨骼肌线粒体自噬蛋白Parkin和分裂蛋白Drp1的表达,增强线粒体融合分裂和自噬能力,且该变化机制部分受到PGC-1α的调控。Verso等[36]研究发现,骨骼肌Atg7-/-小鼠出现严重的线粒体功能受损和氧化应激等症状,而离心收缩运动可加重非功能线粒体的聚集和ROS的产生。并认为,离心收缩损伤刺激时,线粒体质量控制的完整性是维持线粒体功能稳态的重要前提与基础[36]。
这说明,线粒体功能网络的完整性离不开线粒体质量控制,即线粒体生物发生、融合分裂和线粒体自噬等过程。运动既可诱导线粒体生物发生,促进融合分裂循环的动态调整,也可通过线粒体自噬途径回收损伤或衰老的线粒体,产生新的、健康的线粒体,使线粒体进行整体调整和适应,从而维持骨骼肌代谢功能稳态[64](图3)。
首先从产品结构上看,家电市场迎来新一轮的结构调整升级阶段。彩电市场,曲面电视、超轻薄电视的占比持续提升。冰箱市场结构升级加速,多门市场持续爆发,对开门市场也走出价格战的阴霾,风冷向两门和三门产品持续扩散。洗衣机市场,以10KG为代表的大容量滚筒洗衣机引领市场发展,滚筒产品持续挤压波轮产品的份额,而其中的洗烘一体机则成为今年洗衣机市场上最亮眼的黑马。空调市场,柜机的份额持续扩大,而变频柜机则成为市场的利润中心。厨卫电器市场中,近吸式油烟机高速增长以及大火力、高能效、防干烧等节能安全类燃气灶产品的快速增长;小家电市场,破壁料理机成为市场新增长亮点,成长性斐然。

图3 本研究运动对骨骼肌线粒体质量控制的调控机制示意图[28]
3.2 运动训练通过细胞自噬防治代谢相关疾病的分子机制
代谢疾病是由于营养摄取水平改变,能量存储、释放或消耗异常,细胞信号调控机制障碍,激素分泌紊乱等病因导致机体代谢功能失调而诱发的疾病。常见的代谢疾病主要有肥胖、II型糖尿病、肌病、衰老和癌症等。自噬作为骨骼肌中普遍存在的分解代谢调节装置,可清除损伤或衰老的细胞器、ROS等代谢废物,降解糖类、脂类和蛋白质等能源储积物,从而维持骨骼肌代谢稳态,防治代谢疾病发生[45]。目前,有关运动通过骨骼肌细胞自噬防治代谢疾病的研究主要集中于肥胖和II型糖尿病。
骨骼肌是胰岛素依赖的葡萄糖摄取和利用的主要靶器官,也是肥胖和II型糖尿病产生胰岛素抵抗的重要作用部位。自噬在骨骼肌葡萄糖利用和胰岛素抵抗等方面具有重要调节作用。He等[19]研究发现,急性或耐力运动、营养限制可增强野生型小鼠骨骼肌自噬水平,并可显著改善高脂膳食诱导的骨骼肌葡萄糖耐受力下降、瘦素抵抗和高脂血症;但Bcl-2AAA、Beclin1+/-和Atg16l1HM等自噬缺陷小鼠在运动后不仅没有出现上述良性效应,反而加重胰岛素抵抗和糖代谢紊乱等症状,这与运动诱导的骨骼肌葡萄糖摄取、利用和代谢机制障碍有关。这项研究为运动训练通过细胞自噬防治肥胖和II型糖尿病开启了新思路。
肥胖是一种以脂质代谢紊乱为主的代谢疾病,其骨骼肌的主要病理特征是脂滴在胞浆异位储积,并伴有胰岛素抵抗、线粒体功能障碍、内质网应激和葡萄糖耐量下降等症状。许多研究认为,这与肥胖患者营养过剩导致的骨骼肌自噬活性严重削弱有关[12,34,35]。Lv等[37]研究也证实,高糖、高脂膳食均可升高血浆胰岛素水平,导致骨骼肌mTOR的激活,从而削弱自噬活性。而耐力运动则可通过调节骨骼肌自噬水平,改善肥胖患者的相关病理症状。崔迪等[1]研究表明,长期高脂膳食可抑制小鼠骨骼肌ULK1、Atg13和Bnip3/Nix mRNA的表达,降低LC3-II/LC3-I比率,增加P62/SQSTM1蛋白含量,从而下调自噬水平,导致非功能线粒体异常累积,使细胞面临凋亡或坏死风险;而耐力训练则可增加LC3-II蛋白含量,改善Pink1、Parkin和Bnip3/Nix信号转录水平,上调骨骼肌基础自噬和线粒体自噬水平。Fealy等[12]研究认为,肥胖诱导的线粒体质量控制异常是线粒体功能障碍、胰岛素抵抗、甚至II型糖尿病的重要致因,12周耐力训练可下调肥胖型胰岛素抵抗老年人股外侧肌Drp1 Ser616磷酸化水平,促进Opa1和DNM1L/Drp1的表达,并使Mfn1/2、Pink1和Park2表达也略有增加,从而稳定线粒体融合分裂和线粒体自噬,增强胰岛素敏感性和脂肪酸氧化能力[12]。Liu等[34]研究表明,长期高脂膳食可使小鼠血清瘦素、甘油三酯和胆固醇水平升高,葡萄糖耐受力下降,胞浆脂滴过度储积,这与自噬活性减弱有关;耐力训练可逆转高脂膳食诱导的葡萄糖耐受力下降和脂滴过度累积,其与耐力训练时骨骼肌AMPK与Sestrin2/3的相互作用,以及其诱导的基础自噬水平升高密切相关[34]。这说明,耐力训练可重新激活肥胖症骨骼肌细胞自噬和线粒体自噬,降解脂滴和损伤线粒体等易聚集物,改善线粒体功能,抑制糖耐量下降和胰岛素抵抗等症状。
糖尿病是一种在病理机制上与肥胖有着明显区别的能量代谢疾病,但也存在胰岛素抵抗、线粒体功能障碍和骨骼肌流失等症状。Lv等[37]研究表明,链脲佐菌素致II型糖尿病大鼠由于胰岛素缺乏而导致骨骼肌mTOR的抑制和FoxO3的激活,从而使自噬过度激活。Yan等[65]研究发现,GK糖尿病大鼠存在严重的氧化应激、线粒体功能障碍、糖代谢紊乱和肌肉流失等症状,这与LC3-II、Beclin1和Drp1等蛋白过量表达有关;抑制ROS-ERK/JNK-p53信号通路可降低氧化应激,下调自噬水平,改善葡萄糖代谢,抑制骨骼肌过度流失。运动训练也可通过调节自噬通量水平,抑制糖尿病所致的骨骼肌代谢功能紊乱。Lee等[30]研究发现,链脲佐菌素致II型糖尿病大鼠存在骨骼肌质量下降、肌肉萎缩和流失等症状,这与自噬过度激活有关;强迫性游泳训练可显著降低LC3-II蛋白含量,使自噬下调至基础水平,从而减少骨骼肌过度流失。
这说明,运动训练既可上调肥胖症骨骼肌细胞自噬和线粒体自噬水平,降解脂滴、损伤或衰老蛋白质和细胞器,改善糖耐量下降和胰岛素抵抗等症状;也可将糖尿病骨骼肌细胞自噬稳定至基础水平,从而抑制线粒体功能障碍,改善糖代谢紊乱和骨骼肌质量下降等症状。
3.3 运动训练通过细胞自噬维持骨骼肌质量及其功能完整性的分子机制
3.3.1 抗阻运动通过细胞自噬优化骨骼肌质量及其功能的分子机制
骨骼肌质量受到蛋白质合成与降解速率的严密控制与精细调节。一般来说,抗阻运动可刺激骨骼肌蛋白质合成,增加肌肉质量和体积,使骨骼肌产生适应性肥大。抗阻运动诱导的肌肉蛋白质合成受到多条信号通路的调控。其中,PI3K/Akt/mTOR通路是促进肌肉蛋白质合成,维持骨骼肌质量及其功能最经典、最重要的信号通路。
以前研究充分表明,抗阻运动可选择性激活PI3K/Akt/mTOR/S6K1、PI3K/Akt/mTOR/eIF-4E-BP-1/eIF4E、PI3K/Akt/mTOR/GSK-3β/eIF2B等信号通路,促进肌肉蛋白质合成,增加骨骼肌质量和力量。PGC-1α是耐力运动诱导线粒体生物发生的万能转录共激活因子,抗阻运动一般不能通过PGC-1α促进肌肉蛋白质合成。但抗阻运动却可激活PGC-1α的一个重要亚型——PGC-1α4,诱导IGF1结合蛋白的表达,抑制肌肉生长抑制素(myostatin)的基因转录,促进mTOR的激活和S6K1水平的增加,增加肌肉质量和体积[50]。
抗阻运动不仅能刺激肌肉蛋白质合成通路,还能抑制蛋白质降解通路,从而进一步维持骨骼肌质量。FoxO3是一种重要的转录调节因子,可诱导肌萎缩相关基因atrogin-1和MuRF1的表达,通过泛素-蛋白酶体途径降解肌肉蛋白质[56];也可诱导自噬相关基因LC3、Gabarapl1、Bnip3、ULK2、Atg4、Atg 5、Atg 12和Atg 16的表达,通过自噬-溶酶体途径降解肌肉蛋白质[40]。有研究表明,抗阻训练可激活Akt,并磷酸化FoxO3 Thr32/Ser253位点,阻遏其核转位,抑制其介导的两条转录激活机制,减少肌肉蛋白质降解[31]。抗阻训练也可激活PI3K/Akt通路,增强mTORC2的活性,磷酸化抑制FoxO3及其介导的蛋白降解通路。而近期一些研究通过检测FoxO3介导的下游靶基因,进一步证实了抗阻运动介导的Akt/FoxO3通路可下调骨骼肌自噬水平。例如,Fry等[14]研究表明,急性抗阻运动可显著降低骨骼肌LC3-II/LC3-I比率和Gabarap mRNA表达,抑制自噬活性。急性抗阻运动可抑制骨骼肌LC3B-II蛋白表达,下调自噬水平,减少肌肉蛋白质降解[16]。但近期也有研究发现,14天慢性超负荷(抗阻运动)在通过PI3K/Akt/mTOR通路刺激骨骼肌肥大的同时,也可通过mTOR磷酸化ULK1 Ser757位点,抑制ULK1介导的自噬通路,从而下调自噬水平,减少蛋白质降解[58]。这说明,抗阻运动既可激活Akt/FoxO3通路,抑制FoxO3介导的转录激活机制,减少肌肉蛋白质分解;也可激活mTOR/ULK1通路,下调自噬水平,抑制肌肉蛋白质降解。
然而,这种研究结论虽然为解释抗阻运动可抑制肌肉蛋白质降解,从而进一步增加骨骼肌质量提供了良好的证据支持,但却无法解释抗阻运动刺激肌肉收缩时产生的代谢废物(如ROS、损伤或衰老细胞器等)的清除机制。而一些研究也从不同视角证明了自噬在骨骼肌质量维持中的重要作用。骨骼肌Atg7-/-小鼠可出现肌浆网肿胀、损伤线粒体异常聚集和肌节错乱等超微结构紊乱,并存在氧化应激、肌肉萎缩和肌力下降等症状[41]。提示,自噬缺陷不仅不能有效维持骨骼肌质量,反而加重肌肉流失。而骨骼肌Laminin-2-/-小鼠却因自噬过度激活而出现肌肉萎缩和肌营养不良等症状[6]。这说明,过高或过低的自噬水平均可使骨骼肌流失,不益于骨骼肌健康,适度的自噬水平对于维持骨骼肌质量及其功能完整性的意义更加重大。因此,抗阻运动时骨骼肌保持适度的自噬活性或许更能有效维持甚至优化骨骼肌质量及其功能[3,58]。
近期一些研究已经从不同层面论证了抗阻运动诱导骨骼肌细胞自噬的可能分子机制。例如,笔者曾经指出,抗阻运动时骨骼肌中可能存在其他自噬通路(如Beclin1-Vps34通路),且该自噬通路在骨骼肌中可能占据极其重要的地位[3]。最近Neel等[45]也指出,骨骼肌mTOR/ULK1通路仅控制10%的自噬流量,而Akt(Akt/FoxO3和Akt/mTOR通路)可控制50%的自噬流量,其余自噬流量可能由Bcl-2/Beclin1-Vps34[38]、伴侣蛋白辅助选择性自噬(chaperone-assisted selective autophagy,CASA)[62]等通路控制。Mackenzie等[39]研究表明,抗阻运动可使骨骼肌Vps34水平增加,后者既可感受S6K1的变化,也可参与自噬途径中的蛋白质降解。并发现,骨骼肌S6K1在抗阻运动后30 min开始增加,并持续18 h,而Vps34在抗阻运动后3 h开始增加,并维持6 h[39]。从Vps34升高及其与Beclin1的关系可知,Vps34既可作为mTOR上游信号分子刺激肌肉蛋白质合成,也可激活Beclin1-Vps34通路,增加自噬通量水平,降解蛋白质和破损细胞器。该研究组[38]随后发现,抗阻运动在促进蛋白质合成和肌肉质量增加的同时,也部分增加了蛋白质降解,这是通过激活Vps34来实现的。因此,抗阻运动很可能通过Vps34-Beclin1通路诱导细胞自噬,降解胞浆代谢废物。但Mackenzie等[38]同时指出,抗阻运动时Vps34与mTOR介导的蛋白质合成通路的关系可能更密切一些。这说明,抗阻运动通过Beclin1-Vps34通路调节骨骼肌细胞自噬的分子机制仍需进一步研究和证实。
CASA是一种主要回收肌小节细丝蛋白的新型选择性自噬降解途径,其组件蛋白主要包括HSPA8/HSC70、HSPB8/HSP22、BAG3和SYNPO2等[5,62]。CASA可在机械张力刺激下(如抗阻运动)降解细胞骨架蛋白组件,稳定肌肉蛋白质量控制,并在骨骼肌氧化损伤保护和内环境稳态维持等方面发挥重要作用,其功能异常是骨骼肌营养不良和心肌病的重要致因[5]。Ulbricht等[62]研究表明,4周重复性抗阻训练可增加骨骼肌CASA组件蛋白(BAG3、HSPB8、SYNPO2和SQSTM1)表达,增强自噬活性,降解细胞骨架蛋白和损伤细胞器,并产生氨基酸等能量底物,以供合成新的蛋白组件,进而稳定肌肉蛋白质量控制,维持骨骼肌内环境稳态。可知,抗阻训练可激活CASA通路,从而上调自噬水平,降解胞浆中过度累积的代谢废物,进而优化骨骼肌质量及其功能。
尽管多数研究认为抗阻运动可通过多种分子信号途径增加骨骼肌质量和体积,但也有研究并不完全支持运动方式对信号通路的特异性选择,这为抗阻运动通过细胞自噬影响骨骼肌质量提供了新的解释视角。过度负荷在诱导骨骼肌肥大的同时,可适度激活AMPKα1,从而抑制肌肉过度生长[42]。高频电刺激在诱导肌肉收缩的同时,也可激活AMPK,并伴有S6K1、4E-BP1和eEF2表达的降低[61]。抗阻运动可激活AMPK,并降低4E-BP1的磷酸化,抑制肌肉蛋白质过度合成[10]。这说明,过度负荷或高强度剧烈抗阻运动在刺激骨骼肌肥大的同时,也可代偿激活AMPK,其可作为负性调节因子抑制mTOR,阻止骨骼肌过度肥大。由于AMPK是自噬的关键诱导因子,故过度抗阻运动很可能通过AMPK激活细胞自噬,降解胞浆蛋白质或损伤细胞器,从而抑制肌肉过度肥大,巩固和优化骨骼肌质量。
以上研究表明,抗阻运动在增加骨骼肌质量和力量的同时,也可适度上调自噬水平,降解损伤细胞器、ROS和细胞骨架蛋白等胞浆累积物,稳定肌肉蛋白质量控制。因此,抗阻运动诱导的细胞自噬是一种更能有效巩固和优化骨骼肌质量及其功能的重要方式(图4)。
3.3.2 耐力运动通过细胞自噬控制骨骼肌质量及其功能的分子机制
与抗阻运动促进肌肉蛋白质合成、增加骨骼肌质量不同,耐力运动主要增加骨骼肌线粒体含量,改善线粒体功能,增强线粒体有氧代谢,促进肌纤维类型转换。耐力运动也可通过调节多种分子信号途径,抑制肌肉蛋白质合成,甚至增加其降解,产生氨基酸等能量底物,供肌肉收缩或代谢需要[54]。
AMPK作为细胞能量状态的敏感分子,在耐力运动诱导的肌肉蛋白质分解代谢中具有重要作用。以前研究表明,耐力运动、能量匮乏和禁食均可激活骨骼肌AMPK,从而抑制mTOR及其介导的下游蛋白质合成通路,降低骨骼肌质量。但近期研究显示,耐力运动也可通过自噬途径降解非功能或功能性蛋白质、损伤或衰老细胞器,减少骨骼肌质量和体积[47]。耐力运动诱导的AMPK不仅能通过磷酸化激活TSC2、抑制mTOR的方式,减少后者对ULK1的磷酸化,从而上调自噬水平,降解肌肉蛋白质;还能通过直接磷酸化激活ULK1的方式诱导细胞自噬,降解肌肉蛋白质[3]。祖靓等[4]研究表明,力竭耐力运动可提高骨骼肌AMPK的活性和ULK1 Ser317磷酸化水平,增加AMPK与ULK1的结合量,从而增强自噬活性。Moller等[44]研究也证实,耐力运动可激活骨骼肌AMPK,并磷酸化ULK1 Ser555位点,增加ULK1-Atg13-FIP200复合物水平,上调细胞自噬。这说明,耐力运动可通过AMPK/ULK1和AMPK/mTOR通路,诱导细胞自噬,降解肌肉蛋白质和损伤细胞器,降低骨骼肌质量,从而缓解能源物质过度耗竭。
FoxO3及其介导的泛素-蛋白酶体和自噬-溶酶体途径在耐力运动诱导的骨骼肌蛋白质分解代谢中也扮演着核心角色。有研究[21]显示,急性高强度耐力运动可降低骨骼肌Akt、mTOR和4E-BP1水平,抑制Akt介导的FoxO3 Thr32磷酸化;同时促进AMPK的磷酸化,增强FoxO3介导的LC3B-II、Atg4b、Atg12、Bnip3和Cathepsin L表达,降解肌肉蛋白质。这说明,耐力运动可通过激活AMPK/FoxO3通路,降解肌肉蛋白质,产生氨基酸等能量底物,供肌肉收缩和代谢需要,改善有氧耐力水平[21]。Pagano等[47]研究表明,耐力运动激活的AMPK可磷酸化比目鱼肌ULK1 Ser317/Ser555位点,降低Akt Ser473、mTOR Ser2448和4E-BP1 Thr37/Thr346磷酸化水平,抑制mTOR介导的ULK1 Ser757磷酸化和Akt介导的FoxO3a Thr32/Ser253磷酸化,并可增强AMPK/FoxO3a介导的Mul1、MuRF1和LC3B-II表达,降解肌肉蛋白质。提示,耐力运动可通过抑制Akt/mTOR、激活AMPK/ULK1和AMPK/FoxO3a等通路的方式,诱导细胞自噬,促进肌肉蛋白质降解,产生能量底物,供肌肉收缩需要[47]。另外,耐力运动或肌肉收缩刺激也可通过AMPK和P38 MAPK的磷酸化、SIRT1的去乙酰化作用激活PGC-1α,促进FoxO3及其介导的蛋白质水解,降解肌肉蛋白质[13]。禁食或耐力运动时,骨骼肌NAD+/NADH比率升高,从而促进SIRT1依赖的FoxO3去乙酰化激活,降解肌肉蛋白质,产生能量底物[7,13]。但由于FoxO3介导的泛素-蛋白酶体途径可降解约90%的肌肉蛋白质,故自噬的功能可能主要在于清除损伤或衰老细胞器、ROS等代谢废物,维持构成正常水平的骨骼肌蛋白质量控制。
尽管目前多数研究认为耐力运动可降解肌肉蛋白质,减少骨骼肌质量和体积。但近期研究[66]发现,小鼠禁食24 h可降低骨骼肌Akt、S6K1和核糖体S6蛋白激酶(ribosomalS6kinaLse,RSK)的磷酸化水平,抑制mTOR及其介导的蛋白质合成通路,并可提高AMPK的活性和LC3-II/LC3-I比率,诱导细胞自噬;而急性耐力运动却可重新激活禁食小鼠骨骼肌mTOR通路,降低LC3-II/LC3-I比率,下调自噬水平,维持骨骼肌质量及其功能完整性。可知,耐力运动并不总对骨骼肌细胞自噬起正向调节作用,当某些刺激因素导致自噬过度激活时,耐力运动也可下调自噬水平,减少蛋白质或细胞器过度降解,维持骨骼肌代谢稳态。
这说明,耐力运动既可通过多种分子信号途径抑制肌肉蛋白质合成,促进蛋白质水解,并产生氨基酸等能量底物,供肌肉收缩或代谢需要,维持有氧耐力水平;也可通过自噬途径清除胞浆中损伤或衰老细胞器、ROS等代谢废物,维持骨骼肌功能稳态。因此,耐力运动时自噬的主要功能可能并不在于降低骨骼肌质量,而更在于作为一种补偿机制阻止细胞功能流失,保持有氧耐力水平,构成正常水平的肌肉蛋白质量控制以及维持其正常的结构与功能(图4)。

图4 本研究耐力运动和抗阻运动通过细胞自噬调节骨骼肌质量及其功能的分子机制示意图
4 小结与展望
综上所述,细胞自噬作为调节骨骼肌代谢稳态必需的内置机制,可降解肌肉收缩或运动时胞浆中衰老或错误折叠的蛋白质、非功能或损伤细胞组件(线粒体、内质网、核糖体)、病菌、ROS等代谢废物。因此,适量的自噬通量水平可完善骨骼肌线粒体质量控制,稳定线粒体功能网络,维持骨骼肌代谢功能稳态。自噬也可作为一种能源动力系统,降解胞浆中的能源储积物(糖原、脂质和蛋白质等),产生葡萄糖、自由脂肪酸和氨基酸等能量底物,提供细胞更新和代谢平衡所需的能量与合成底物。因此,自噬也可有效防治胰岛素抵抗、肥胖和II型糖尿病等代谢疾病发生。骨骼肌质量及其功能完整性是稳定骨骼肌代谢稳态的重要保证。抗阻运动不仅能增加骨骼肌质量和力量,还能通过其介导的自噬进一步巩固和优化骨骼肌质量;耐力运动介导的自噬的主要功能可能并不在于降低骨骼肌质量,而更在于作为一种补偿机制阻止细胞功能流失,保持有氧耐力水平,维持构成正常水平的肌肉蛋白质量控制,以及稳定骨骼肌代谢功能稳态。
但目前有关运动训练通过细胞自噬调节骨骼肌代谢稳态的分子信号机制还存在一些亟待进一步研究和探讨的重要问题:1)自噬是一种具有高度特异选择性的降解途径,主要包括线粒体自噬、内质网自噬和核糖体自噬等。目前多数研究均聚焦于运动诱导的线粒体自噬在稳定线粒体质量控制、维持骨骼肌代谢稳态中的重要作用。有关内质网自噬和核糖体自噬的降解机制和潜在事件却知之甚少,而长期规律性运动可否使内质网自噬和核糖体自噬更好的适应蛋白翻译机制也亟待探寻。2)尽管目前多数研究认为,运动介导的细胞自噬在骨骼肌葡萄糖利用和胰岛素抵抗等方面发挥重要作用。但近期却有研究[26]发现,骨骼肌Atg7-/-小鼠瘦体重和脂肪储量降低,葡萄糖耐受力、胰岛素敏感性和能量消耗提高,即使经过高脂膳食也未出现肥胖和胰岛素抵抗,这源于自噬缺陷时骨骼肌内分泌信号的代偿分泌与释放,以及其诱导的脂解作用和脂肪酸β氧化。这与之前的研究结果存在一定的矛盾,其具体原因也有待深入探讨。3)抗阻运动通过Beclin1-Vps34和CASA等通路诱导细胞自噬的分子信号机制,以及过度抗阻运动通过代偿激活AMPK及其介导的细胞自噬,从而巩固和优化骨骼肌质量及其功能的分子机制也需要进一步研究。深入探析和解决这些问题,将会更加充分地揭示运动介导的细胞自噬在调节骨骼肌代谢稳态中的重要作用。
[1]崔迪,邱守涛,王海燕,等.耐力运动对营养性肥胖小鼠骨骼肌细胞自噬及线粒体自噬的影响[J].体育科学,2014,34(12):63-71.
[2]漆正堂,郭维,张媛,等.不同运动方式对大鼠骨骼肌线粒体融合分裂基因及Mfn2、Drp1蛋白表达的影响[J].中国运动医学杂志,2011,30(2):143-148.
[3]钱帅伟,罗艳蕊,漆正堂,等.细胞自噬的分子学机制及运动训练的调控作用[J].体育科学,2012,32(1):64-70.
[4]祖靓,朱荣.力竭运动对小鼠骨骼肌细胞自噬的影响及相关调节机制研究[J].体育科学,2013,33(9):77-84.
[5]ARNDT V,DICK N,TAWO R,etal.Chaperone-assisted selective autophagy is essential for muscle maintenance[J].Curr Biol,2010,20(2):143-148.
[6]BONALDO P,SANDRI M.Cellular and molecular mechanisms of muscle atrophy[J].Dis Model Mech,2013,6(1):25-39.
[7]CANTO C,GERHART-HINES Z,FEIGE J N,etal.AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity[J].Nature,2009,458(7241):1056-1060.
[8]COBLEY J N,BARTLETT J D,KAYANI A,etal.PGC-1alpha transcriptional response and mitochondrial adaptation to acute exercise is maintained in skeletal muscle of sedentary elderly males[J].Biogerontology,2012,13(6):621-631.
[9]DAVIDSON L E,HUDSON R,KILPATRICK K,etal.Effects of exercise modality on insulin resistance and functional limitation in older adults:a randomized controlled trial[J].Arch Intern Med,2009,169(2):122-131.
[10]DREYER H C,FUJITA S,CADENAS J G,etal.Resistance exercise increases AMPK activity and reduces 4E-BP1 phosphorylation and protein synthesis in human skeletal muscle[J].J Physiol,2006,576(Pt 2):613-624.
[11]EGAN B,ZIERATH J R.Exercise metabolism and the molecular regulation of skeletal muscle adaptation[J].Cell Metab,2013,17(2):162-184.
[12]FEALY C E,MULYA A,LAI N,etal.Exercise training decreases activation of the mitochondrial fission protein dynamin-related protein-1 in insulin-resistant human skeletal muscle[J].J Appl Physiol (1985),2014,117(3):239-245.
[13]FERRARO E,GIAMMARIOLI A M,CHIANDOTTO S,etal.Exercise-induced skeletal muscle remodeling and metabolic adaptation:redox signaling and role of autophagy[J].Antioxid Redox Signal,2014,21(1):154-176.
[14]FRY C S,DRUMMOND M J,GLYNN E L,etal.Skeletal muscle autophagy and protein breakdown following resistance exercise are similar in younger and older adults[J].J Gerontol A Biol Sci Med Sci,2013,68(5):599-607.
[15]GARNIER A,FORTIN D,ZOLL J,etal.Coordinated changes in mitochondrial function and biogenesis in healthy and diseased human skeletal muscle[J].FASEB J,2005,19(1):43-52.
[16]GLYNN E L,FRY C S,DRUMMOND M J,etal.Muscle protein breakdown has a minor role in the protein anabolic response to essential amino acid and carbohydrate intake following resistance exercise[J].Am J Physiol Regul Integr Comp Physiol,2010,299(2):R533-R540.
[17]GRUMATI P,COLETTO L,SCHIAVINATO A,etal.Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI-deficient muscles[J].Autophagy,2011,7(12):1415-1423.
[18]GUO W,WONG S,LI M,etal.Testosterone plus low-intensity physical training in late life improves functional performance,skeletal muscle mitochondrial biogenesis,and mitochondrial quality control in male mice[J].PLoS One,2012,7(12):e51180.
[19]HE C,BASSIK M C,MORESI V,etal.Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis[J].Nature,2012,481(7382):511-515.
[20]HUDSON M B,RAHNERT J A,ZHENG B,etal.miR-182 attenuates atrophy-related gene expression by targeting FoxO3 in skeletal muscle[J].Am J Physiol Cell Physiol,2014,307(4):C314-C319.
[21]JAMART C,FRANCAUX M,MILLET G Y,etal.Modulation of autophagy and ubiquitin-proteasome pathways during ultra-endurance running[J].J Appl Physiol (1985),2012,112(9):1529-1537.
[22]JAMART C,NASLAIN D,GILSON H,etal.Higher activation of autophagy in skeletal muscle of mice during endurance exercise in the fasted state[J].Am J Physiol Endocrinol Metab,2013,305(8):E964-E974.
[23]KANG R,ZEH H J,LOTZE M T,etal.The Beclin1 network regulates autophagy and apoptosis[J].Cell Death Differ,2011,18(4):571-580.
[24]KIM J,KIM Y C,FANG C,etal.Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy[J].Cell,2013,152(1-2):290-303.
[25]KIM J,KUNDU M,VIOLLET B,etal.AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1[J].Nat Cell Biol,2011,13(2):132-141.
[26]KIM K H,JEONG Y T,OH H,etal.Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine[J].Nat Med,2013,19(1):83-92.
[27]KIM K H,LEE M S.Autophagy as a crosstalk mediator of metabolic organs in regulation of energy metabolism[J].Rev Endocr Metab Disord,2014,15(1):11-20.
[28]KLUGE M A,FETTERMAN J L,VITA J A.Mitochondria and endothelial function[J].Circ Res,2013,112(8):1171-1188.
[29]LEE I H,FINKEL T.Regulation of autophagy by the p300 acetyltransferase[J].J Biol Chem,2009,284(10):6322-6328.
[30]LEE Y,KIM J H,HONG Y,etal.Prophylactic effects of swimming exercise on autophagy-induced muscle atrophy in diabetic rats[J].Lab Anim Res,2012,28(3):171-179.
[31]LEGER B,CARTONI R,PRAZ M,etal.Akt signalling through GSK-3beta,mTOR and Foxo1 is involved in human skeletal muscle hypertrophy and atrophy[J].J Physiol,2006,576(Pt 3):923-933.
[32]LIRA V A,OKUTSU M,ZHANG M,etal.Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance[J].FASEB J,2013,27(10):4184-4193.
[33]LITTLE J P,SAFDAR A,BISHOP D,etal.An acute bout of high-intensity interval training increases the nuclear abundance of PGC-1alpha and activates mitochondrial biogenesis in human skeletal muscle[J].Am J Physiol Regul Integr Comp Physiol,2011,300(6):R1303-R1310.
[34]LIU X,NIU Y,YUAN H,etal.AMPK binds to Sestrins and mediates the effect of exercise to increase insulin-sensitivity through autophagy[J].Metabolism,2015,64(6):658-665.
[35]LIU Y,PALANIVEL R,RAI E,etal.Adiponectin stimulates autophagy and reduces oxidative stress to enhance insulin sensitivity during high-fat diet feeding in mice[J].Diabetes,2015,64(1):36-48.
[36]LO V F,CARNIO S,VAINSHTEIN A,etal.Autophagy is not required to sustain exercise and PRKAA1/AMPK activity but is important to prevent mitochondrial damage during physical activity[J].Autophagy,2014,10(11):1883-1894.
[37]LV P,HUANG J,YANG J,etal.Autophagy in muscle of glucose-infusion hyperglycemia rats and streptozotocin-induced hyperglycemia rats via selective activation of m-TOR or FoxO3[J].PLoS One,2014,9(2):e87254.
[38]MACKENZIE M G,HAMILTON D L,MURRAY J T,etal.mVps34 is activated following high-resistance contractions[J].J Physiol,2009,587(Pt 1):253-260.
[39]MACKENZIE M G,HAMILTON D L,MURRAY J T,etal.mVps34 is activated by an acute bout of resistance exercise[J].Biochem Soc Trans,2007,35(Pt 5):1314-1316.
[40]MAMMUCARI C,MILAN G,ROMANELLO V,etal.FoxO3 controls autophagy in skeletal muscle in vivo[J].Cell Metab,2007,6(6):458-471.
[41]MASIERO E,SANDRI M.Autophagy inhibition induces atrophy and myopathy in adult skeletal muscles[J].Autophagy,2010,6(2):307-309.
[42]MCGEE S L,MUSTARD K J,HARDIE D G,etal.Normal hypertrophy accompanied by phosphoryation and activation of AMP-activated protein kinase alpha1 following overload in LKB1 knockout mice[J].J Physiol,2008,586(6):1731-1741.
[43]MCMILLAN E M,PARE M F,BAECHLER B L,etal.Autophagic signaling and proteolytic enzyme activity in cardiac and skeletal muscle of spontaneously hypertensive rats following chronic aerobic exercise[J].PLoS One,2015,10(3):e119382.
[44]MOLLER A B,VENDELBO M H,CHRISTENSEN B,etal.Physical exercise increases autophagic signaling through ULK1 in human skeletal muscle[J].J Appl Physiol (1985),2015,118(8):971-979.
[45]NEEL B A,LIN Y,PESSIN J E.Skeletal muscle autophagy:a new metabolic regulator[J].Trends Endocrinol Metab,2013,24(12):635-643.
[46]NEMAZANYY I,BLAAUW B,PAOLINI C,etal.Defects of Vps15 in skeletal muscles lead to autophagic vacuolar myopathy and lysosomal disease[J].EMBO Mol Med,2013,5(6):870-890.
[47]PAGANO A F,PY G,BERNARDI H,etal.Autophagy and protein turnover signaling in slow-twitch muscle during exercise[J].Med Sci Sports Exe,2014,46(7):1314-1325.
[48]QI Z,ZHANG Y,GUO W,etal.Increased insulin sensitivity and distorted mitochondrial adaptations during muscle unloading[J].Int J Mol Sci,2012,13(12):16971-16985.
[49]REYNOLDS T T,MERRELL E,CINQUINO N,etal.Disassociation of insulin action and Akt/FOXO signaling in skeletal muscle of older Akt-deficient mice[J].Am J Physiol Regul Integr Comp Physiol,2012,303(11):R1186-R1194.
[50]RUAS J L,WHITE J P,RAO R R,etal.A PGC-1alpha isoform induced by resistance training regulates skeletal muscle hypertrophy[J].Cell,2012,151(6):1319-1331.
[51]RUSSELL A P,FOLETTA V C,SNOW R J,etal.Skeletal muscle mitochondria:a major player in exercise,health and disease[J].Biochim Biophys Acta,2014,1840(4):1276-1284.
[52]RUSSELL R C,TIAN Y,YUAN H,etal.ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase[J].Nat Cell Biol,2013,15(7):741-750.
[53]SALMINEN A,VIHKO V.Autophagic response to strenuous exercise in mouse skeletal muscle fibers[J].Virchows Arch B Cell Pathol Incl Mol Pathol,1984,45(1):97-106.
[54]SANCHEZ A M,BERNARDI H,PY G,etal.Autophagy is essential to support skeletal muscle plasticity in response to endurance exercise[J].Am J Physiol Regul Integr Comp Physiol,2014,307(8):R956-R969.
[55]SANCHEZ A M,CSIBI A,RAIBON A,etal.AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1[J].J Cell Biochem,2012,113(2):695-710.
[56]SANDRI M,SANDRI C,GILBERT A,etal.Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy[J].Cell,2004,117(3):399-412.
[57]SETTEMBRE C,ZONCU R,MEDINA D L,etal.A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB[J].EMBO J,2012,31(5):1095-1108.
[58]STEINER J L,GORDON B S,LANG C H.Moderate alcohol consumption does not impair overload-induced muscle hypertrophy and protein synthesis[J].Physiol Rep,2015,3(3):e12333.
[59]SUWA M,NAKANO H,RADAK Z,etal.Endurance exercise increases the SIRT1 and peroxisome proliferator-activated receptor gamma coactivator-1alpha protein expressions in rat skeletal muscle[J].Metabolism,2008,57(7):986-998.
[60]TAM B T,SIU P M.Autophagic cellular responses to physical exercise in skeletal muscle[J].Sports Med,2014,44(5):625-640.
[61]THOMSON D M,FICK C A,GORDON S E.AMPK activation attenuates S6K1,4E-BP1,and eEF2 signaling responses to high-frequency electrically stimulated skeletal muscle contractions[J].J Appl Physiol(1985),2008,104(3):625-632.
[62]ULBRICHT A,GEHLERT S,LECIEJEWSKI B,etal.Induction and adaptation of chaperone-assisted selective autophagy CASA in response to resistance exercise in human skeletal muscle[J].Autophagy,2015,11(3):538-546.
[63]VAINSHTEIN A,TRYON L D,PAULY M,etal.The role of PGC-1alpha during acute exercise-induced autophagy and mitophagy in skeletal muscle[J].Am J Physiol Cell Physiol,2015,308(9):C710-C719.
[64]WIGGS M P.Can endurance exercise preconditioning prevention disuse muscle atrophy?[J].Front Physiol,2015,6:63.
[65]YAN J,FENG Z,LIU J,etal.Enhanced autophagy plays a cardinal role in mitochondrial dysfunction in type 2 diabetic Goto-Kakizaki(GK)rats:ameliorating effects of (-)-epigallocatechin-3-gallate[J].J Nutr Biochem,2012,23(7):716-724.
[66]ZHENG D M,BIAN Z,FURUYA N,etal.A treadmill exercise reactivates the signaling of the mammalian target of rapamycin (mTor) in the skeletal muscles of starved mice[J].Biochem Biophys Res Commun,2015,456(1):519-526.
Exercise-mediated Autophagy is a Built-in Mechanism to Regulate Skeletal Muscle Metabolic Homeostasis
QIAN Shuai-wei1,2,DING Shu-zhe1
As a compensatory and built-in mechanism of skeletal muscle,autophagy not only could degrade reactive oxygen species,bacteria,aging or damaged organelles such as mitochondria,endoplasmic reticulum,ribosome,as well as degrade glycogen,lipid,non-functional and functional protein when suffer energy stress such as exercise,fasting,nutrition restriction and muscle contraction.Autophagy accordingly could improve muscle quality control,as well as energy and synthetic substrates for cellular renewal and metabolism.Exercise training-mediated autophagy not only could improve mitochondrial quality control,stabilize mitochondrial function network,as well as maintain metabolic homeostasis of muscle,but also effectively prevent insulin resistance,obesity,type II diabetes and some other metabolic diseases.Exercise training-mediated autophagy could also entirely adapt muscle mass and function to their items,and further improve muscle metabolism and functional homeostasis.
autophagy;skeletalmuscle;exercisetraining;metabolichomeostasis;mitochondrialqualitycontrol;metabolicdiseases;musclemass
2015-07-28;
2015-09-21
国家自然科学基金资助项目(31171142)。
钱帅伟(1984-),男,河南漯河人,讲师,在读博士研究生,主要研究方向为运动通过细胞自噬防治肥胖和II型糖尿病的分子信号机制,E-mail:qianshuaiwei999@163.com;丁树哲(1963-),男,黑龙江望奎人,教授,博士,博士研究生导师,主要研究方向为运动适应与线粒体信号调控,E-mail:szding@tyxx.ecnu.edu.cn,Tel:(021)62235425。
1.华东师范大学 青少年健康评价与运动干预教育部重点实验室,上海 200241;2.烟台大学 体育学院,山东 烟台 264005 1.East China Normal University,Shanghai 200241,China;2.Yantai University,Yantai 264005,China.
1000-677X(2015)10-0055-11
10.16469/j.css.201510008
G804.5
A
