Brain protein oxidation: what does it refl ect?
2015-02-07ParvanaHajieva,BerndMoosmann
Brain protein oxidation: what does it refl ect?
Antioxidant neuroprotection: Since the elaboration of the concept of oxidative stress in the 1980s, the idea that this phenomenon may be particularly involved in diseases of the brain has become widely accepted (Halliwell, 2006). Embedded in the framework of neuroprotection, the investigation of antioxidant strategies was fuelled by the repeated observation of redox dysregulation and outright oxidative damage on the molecular scale in many chronic and acute conditions involving neuronal dysfunction (Moosmann and Behl, 2002). In fact, diff erent approaches of pharmacological antioxidant neuroprotection worked surprisingly well in animal studies; however, they have so far refused to work, almost without exception, in the clinic. The failure of NXY-059 in 2007, which was the latest candidate in a series of substances tested for ischemic stroke, was a disturbing setback in this respect (Shuaib et al., 2007). The very obvious discrepancy between success rates in mice, rats and humans had not been anticipated, as many drugs based neuronal receptor pharmacology had found their ready translation from animal studies into the clinic. What might have been the specifi c causes of failure when it comes to antioxidant neuroprotection?
Clinical issues: The three most frequently cited answers may be summarized as (i) “chemical failure”, (ii) “technical failure”, and (iii) “biological failure”. Answer (i) claims that insufficient basic drug effi cacy in terms of a high EC50value or inadequate bloodbrain barrier permeability was causative, answer (ii) argues that the drugs were satisfying but that technical hurdles such as temporally later administration in clinical settings compared to animal studies or more heterogeneous treatment populations were to be blamed, and answer (iii) predicates that both of the above were less relevant than the insuffi cient knowledge about disease causalities and the biological responses of the body to the drug. There may have been, for example, an adaptive downregulation of endogenous antioxidant defenses or other dynamic biological changes leaving no room for the accrual of a net benefi t. Reasonable evidence has been provided for each of these alternatives in one or the other disease model. Still, what has seemingly never been investigated prior to our recent study (Granold et al., 2015) is the possibility that mice, rats and humans may, in some unknown respect, be intrinsically diff erent in terms of their baseline patterns of oxidative damage.
Brain protein oxidation: Starting in on protein oxidation as a case in point, we performed a direct inter-species comparison of the baseline levels of membrane protein oxidation and cytosolic protein oxidation in mice, rats, and humans, taking lipid peroxidation as a reference marker. As expected, we usually found that baseline levels of oxidative damage were much lower in long-lived humans than in short-lived rodents. This observation applied to both markers 8-isoprostane immunoreactivity and protein carbonyl chemoreactivity in cytosolic proteins in cortical as well as cerebellar tissue. To our surprise, though, membrane protein oxidation in the human cerebral cortex appeared to be detached from this largely consistent picture, as we detected the highest levels of damage of all specimens in this fraction. Hence, the carbonyl content of human cortical membrane proteins exceeded that of mouse cortical membrane proteins or human cerebellar membrane proteins, despite the fact that lipid peroxidation and cytosolic protein oxidation in the same samples were utterly low. How to explain such a result in markers of oxidation that are often considered equivalent in mice and humans?
Membrane proteins: From a structural point of view, membrane proteins might be particularly exposed to reactive oxygen species as they are immersed into the membrane, in which peroxyl radicals emerging from chain reactions are much more concentrated than in the aqueous space, especially under pathological conditions (Hajieva et al., 2015). While this structural interpretation might clearly contribute to the answer as it correctly predicts a diff erence between membrane and cytosol, it leaves unresolved why humans, and within humans, why cortex is primarily aff ected. Evidently, there is little room for any speculation that higher exposure to oxidants might also explain the species diff erence, as to all knowledge, humans generate much lower fl uxes of oxidants than rodents (Kudin et al., 2008), which is concordant with our fi nding of very low lipid peroxidation and cytosolic protein oxidation (Granold et al., 2015). A major part of the answer might rather come from a diff erent direction, namely from the consideration that steady-state levels of macromolecular oxidative damage necessarily refl ect exposure (per time), repair (per time), and lifetime. As protein carbonyls are most likely not repaired, the question arises whether there may exist substantial diff erences in the brains of mice and men regarding protein longevity. Could higher steady-state levels of oxidation actually refl ect longer protein half-lives, either in a functional or dysfunctional state?
Protein turnover: Mouse liver proteins have an average lifetime of about 3 days, with large variation, while mouse brain proteins persist signifi cantly longer and reach mean lifetimes of about 9 days (Price et al., 2010). Notably, the longest-lived classes of proteins in both tissues are polytopic membrane proteins. In the brain, proteins from the myelin sheath, the nuclear membrane, and the inner mitochondrial membrane are among the longest-lived examples and can reach lifetimes of several weeks. Hence, membrane proteins are indeed longer-lived than cytosolic proteins, but this diff erence seems to entail only a minor bias towards higher membrane protein oxidation in mice (Granold et al., 2015). The relevant point that needs to be addressed in consequence, but which cannot be answered satisfactorily to date, is whether proteins in the human cortex have similar, or possibly much longer lifetimes than their mouse counterparts. Evolutionary biology would argue that larger animals like humans have lower metabolic rates and in connection, lower protein turnover, involving longer molecular lifetimes. However, brain metabolic rates in mice and humans are actually very similar and thus constitute an exception from the rule (Aiello and Wheeler, 1995). Moreover, the pronounced neocortical size expansion that has taken place within just a few million years in humans (Figure 1) may have been too rapid to enable the adaptive structural evolution of all cortical proteins to acquire suffi cient stability to result in a proportionally increased lifetime. What would happen, thus, if longer lifetimes were forcefully imposed on the brain’s proteins by human brain anatomy even if they were not yet adapted structurally to such extended lifetimes?
Axonal transport: It is palpable from plain anatomy that many human cortical proteins must fulfi ll an unusual demand regarding their lifetime, namely stability during extended periods of axonal transport. In the human cortex, the average spatial distance between functionally connected sites has been estimated to be in the range of 70,000 μm (Bullmore and Bassett, 2011). Assuming direct proportionality with cortical size, the corresponding distance in mice would be approximately 3,000 μm (Figure 1). Hence, it is at least these (Euclidean) distances that proteins synthesized and assembled perinuclearly have to travel to reach their presynaptic site of function, and many of them will have to travel back the same distance for fi nal disposal. As there is no indication that axonal transport is much faster in humans than in mice, synaptic vesicles, generally thought to be the fastest travelling structures (velocity 1.2–1.5 μm/s), will be on the road for a minimum of about 0.5 days in humans, whereas membrane protein-loaded mitochondria probably travel for at least 1.5–4 days (velocity 0.2–0.6 μm/s) (MacAskill and Kittler, 2010). Homologous mouse proteins will be travelling for only about 1/25 of these intervals. Thus, transport times appear to be negligible in mice, but relevant in humans, especially if one assumes that both species might have comparable protein lifetimes(of about 9 days). However, it is quite likely from fi rst principles that human proteins travelling anterogradely for 1.5–4 days will not exhibit lifetimes of merely 9 days, such that for many membrane proteins, signifi cantly longer lifetimes need to apply. Hence, if proteins evolved for relatively short lifetimes were rather suddenly obliged to survive long transport times during which stability and oxidative damage may not be as stringently surveilled as at the synapse, it is quite well conceivable that these proteins might suff er selectively from increased oxidation.
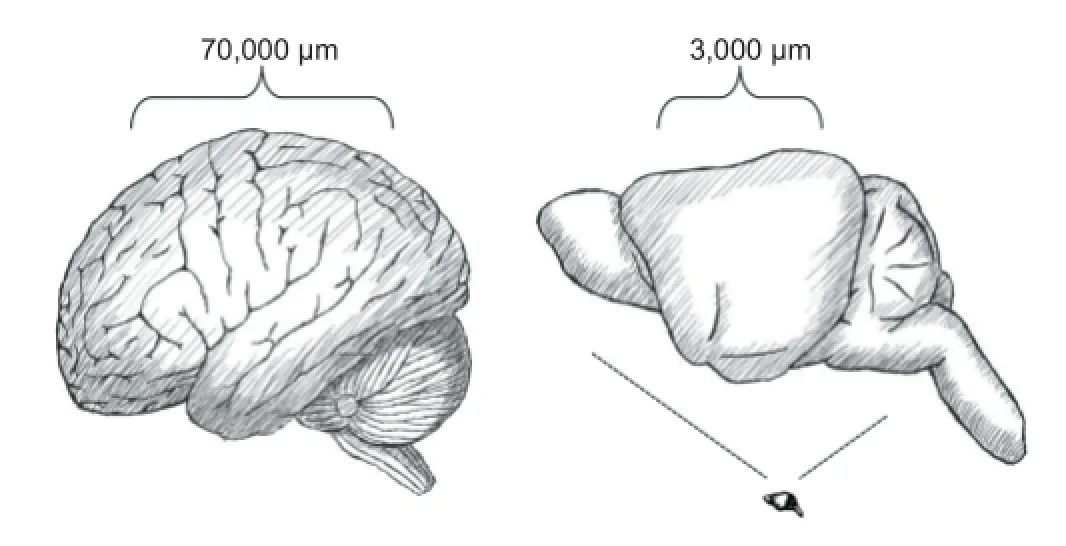
Figure 1 Illustration of the average distances of functionally connected sites in the cerebral cortex of humans (70,000 μm, left) and mice (3,000 μm, right).
Protein degradation: An even more compelling argument might still arise from retrograde axonal transport. Notably, cytosolic proteins in the cortex are readily degraded locally, by the dendritically and axonally ubiquitous proteasomes (Tai and Schuman, 2008). In fact, it has been shown that protein carbonyls serve as signals for degradation by the 20S proteasome, at least in cytosolic and moderately hydrophobic proteins (Höhn et al., 2013). In contrast, this option seems to be blocked for the severely hydrophobic membrane proteins, as there is clear evidence of retrograde transport of multivesicular bodies, aged mitochondria and even autophagosomes all the way down to the perinuclear space at a mean velocity of about 0.45 μm/s in vitro (Maday et al., 2012). This velocity would translate into an average travelling time of about another 2 days in vivo after the “decision to degrade”. Was the latter decision based on exceeding oxidation, these oxidized structures would persist without further maintenance or repair and would be detectable for at least the necessary transport time. Besides, more than 25% of autophagosomes seem to travel at 0.1 μm/s or less, implying a travelling time of more than 8 days if these structures were indeed a separate class of cargo (Maday et al., 2012). And throughout retrograde transport, these oxidized and damaged hydrophobic proteins might start to aggregate or display other types of toxic gain of function. At present, it appears that long retrograde travelling times of oxidized membrane proteins already marked for degradation provide one of the best explanations for the experimentally observed pattern of high protein oxidation limited to humans, to cortex, and to membrane proteins. In addition, two rather odd observations from animal studies might be much easier to explain when the peak markers of protein oxidation in the brain were primarily related to structures that are not under redox surveillance anymore. First, the fact that acute antioxidants often provide benefi t in vivo even if they do not lower the predominant markers of oxidation. Concordantly, they sometimes help in models that do not even display elevated peak markers of oxidation. Second, the fact that protein oxidation shows considerable inter-individual variability, while within each individual, rather small increases on top of the individual baseline suffi ce to cause cellular damage.
Conclusion: Granted that the “oxidation-through-lifetime” hypothesis was correct, what consequences would emerge for future neuroprotective strategies? Most basically, the realization that humans and mice are diff erent, even with respect to such fundamental aspects of redox homeostasis as protein carbonyl formation. In humans, cytosolic protein oxidation appears to be less of a problem than damage to membrane proteins, which might account for the failure in humans of the exclusively aqueous, double-sulfo compound NXY-059 that had been quite effi cacious in rodents. Gyrencephalic animal models might provide some solution to avoid the rodent-human gap, as proposed by the Stroke Therapy Academic Industry Roundtable (STAIR) recommendations for stroke, but it is clear that those models are very much demanding. What could one still achieve in mice? Clearly, the study of protein turnover should provide signifi cant insight. Moreover, the identifi cation of proteins with very high baseline levels of oxidation, or the search for proteins that are selectively degraded after an insult or in a disease might be rewarding. Much less rewarding might be the further search for proteins whose steady-state levels of oxidation are somewhat higher in a disease than in healthy controls: those proteins might just represent a subset of proteins whose oxidation is extraordinarily well tolerated by the cell, such that the cell postpones their degradation to a later point (Granold et al., 2015). In the end, the study of protein oxidation in the dynamic context of turnover and site-specifi c degradation in the brain seems crucial, particularly for the development of better treatment options for those neurological disorders that are caused or strongly infl uenced by proteolytic failure: the Neuronal Ceroid Lipofuscinoses (NCL), or Parkinson’s disease (PD), to name two prominent examples.
Parvana Hajieva, Bernd Moosmann*
Institute for Pathobiochemistry, University Medical Center of the Johannes Gutenberg University, Mainz, Germany
*Correspondence to: Bernd Moosmann, Ph.D., moosmann@uni-mainz.de.
Accepted: 2015-07-15
Aiello LC, Wheeler P (1995) The expensive-tissue hypothesis: the brain and the digestive system in human and primate evolution. Curr Anthropol 36:199-221.
Bullmore ET, Bassett DS (2011) Brain graphs: graphical models of the human brain connectome. Annu Rev Clin Psychol 7:113-140.
Granold M, Moosmann B, Staib-Lasarzik I, Arendt T, Del Rey A, Engelhard K, Behl C, Hajieva P (2015) High membrane protein oxidation in the human cerebral cortex. Redox Biol 4:200-207.
Hajieva P, Bayatti N, Granold M, Behl C, Moosmann B (2015) Membrane protein oxidation determines neuronal degeneration. J Neurochem 133:352-367. Halliwell B (2006) Oxidative stress and neurodegeneration: where are we now? J Neurochem 97:1634-1658.
Höhn A, König J, Grune T (2013) Protein oxidation in aging and the removal of oxidized proteins. J Proteomics 92:132-159.
Kudin AP, Malinska D, Kunz WS (2008) Sites of generation of reactive oxygen species in homogenates of brain tissue determined with the use of respiratory substrates and inhibitors. Biochim Biophys Acta 1777:689-695.
MacAskill AF, Kittler JT (2010) Control of mitochondrial transport and localization in neurons. Trends Cell Biol 20:102-112.
Moosmann B, Behl C (2002) Antioxidants as treatment for neurodegenerative disorders. Expert Opin Investig Drugs 11:1407-1435.
Price JC, Guan S, Burlingame A, Prusiner SB, Ghaemmaghami S (2010) Analysis of proteome dynamics in the mouse brain. Proc Natl Acad Sci U S A 107:14508-14513.
Shuaib A, Lees KR, Lyden P, Grotta J, Davalos A, Davis SM, Diener HC, Ashwood T, Wasiewski WW, Emeribe U, SAINT II Trial Investigators (2007) NXY-059 for the treatment of acute ischemic stroke. N Engl J Med 357:562-571.
Tai HC, Schuman EM (2008) Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci 9:826-838.
10.4103/1673-5374.170294 http://www.nrronline.org/
Hajieva P, Moosmann B (2015) Brain protein oxidation: what does it refl ect? Neural Regen Res 10(11):1729-1730.
杂志排行

中国神经再生研究(英文版)的其它文章
- Intracellular sorting pathways of the amyloid precursor protein provide novel neuroprotective strategies
- The role of the Rho/ROCK signaling pathway in inhibiting axonal regeneration in the central nervous system
- VEGF in the nervous system: an important target for research in neurodevelopmental and regenerative medicine
- Studying neurological disorders using induced pluripotent stem cells and optogenetics
- Ef cacy of glucagon-like peptide-1 mimetics for neural regeneration
- Compliant semiconductor scaf olds: building blocks for advanced neural interfaces