一种基于线性DNA片段同源重组的嗜盐古菌高效基因敲除系统
2015-02-04王小利姜闯刘建华刘喜朋
王小利,姜闯,刘建华,刘喜朋
一种基于线性DNA片段同源重组的嗜盐古菌高效基因敲除系统
王小利,姜闯,刘建华,刘喜朋
上海交通大学生命科学技术学院,微生物代谢国家重点实验室, 上海 200240
随着功能基因组学研究的深入发展,基因敲除技术日益成为基因功能研究的重要手段。嗜盐古菌易于培养,是研究古菌基因功能的良好模式菌株。虽然现已开发了多种嗜盐古菌的遗传操作系统,但基因敲除成功率不十分理想。这些遗传操作方法基于筛选标记,利用携带同源片段的环状质粒与基因组同源片段间的两次同源重组,敲除目的基因。由于基于环状质粒和筛选标记的经典同源重组敲除方法在二次重组时,普遍存在回复到野生型菌株的可能,导致二次重组子中敲除目的基因的阳性菌株比例较低。为了克服传统同源重组技术的上述缺陷,文章建立了基于线性DNA片段的同源重组技术。该方法通过一次重组在目标基因的下游引入一段上游同源片段和标记,从而限定二次重组的发生部位只能在两段上游同源片段之间,发生二次重组的重组子理论上都敲除了目标基因。利用该方法,文章成功敲除了嗜盐古菌的基因,阳性克隆率达65%。这种线性DNA片段重组法为嗜盐古菌的基因敲除提供了一种高效策略,便于嗜盐古菌的基因改造。
嗜盐古菌;基因敲除;同源重组;线性DNA重组
遗传操作系统是基因敲除等分子遗传学研究的基础,为了研究目的基因敲除后的各种表型,首先需要获得目的基因敲除突变株。目前已在多种生物中建立了敲除目的基因的遗传操作系统。为了加速认识古菌的基因功能,目前成功建立了针对多种古菌的遗传操作系统[1~4]。古菌传统遗传操作系统是基于尿嘧啶营养缺陷型(∆)和5-氟乳清酸抗性(5-FOA)的正负筛选标记的pop-in/pop-out 基因敲除策略,环状载体的一侧同源臂与染色体上的同源区发生一次重组,导致载体整合到染色体中,即pop-in,尿嘧啶营养缺陷型菌株恢复为原养型。然后,通过分子内重组,将带有的载体去除,即pop-out,丢掉基因的菌株可以在含5-FOA的培养基中生长。在5-FOA平板生长的菌株有两种基因型:靶基因缺失及靶基因未缺失(即恢复到野生型),需要通过分子杂交或PCR区分两种基因型菌株[5,6]。目前具有相对成熟遗传操作系统的古菌包括:沃氏嗜盐富饶菌()[5,6]、地中海富盐菌()[7]、西班牙盐杆菌()[7,8]、嗜酸热硫化叶菌()[9]、冰岛硫化叶菌()[10]、激烈火球菌()[11]和栖热球菌()[12]等。
嗜盐古菌分离自死海,属于古菌域广古菌门嗜盐富饶菌属(),细胞为不规则的杆状、盘状或杯状(1.0~3.0 μm×2.0~3.0μm),具有鞭毛,可以运动。在实验室环境易于培养,最适培养条件为:18%盐水培养基、温度45℃。由于该菌是易于培养的好氧古菌,并已完成全基因组测序[13],便于基因敲除操作,因此是研究古菌基因功能的良好模式菌株[2]。目前,已建立成熟高效的转化系统[14],现有基因敲除系统具有高效标记基因[15,16]用于筛选突变株,并开发了多种表达载体[17,18]。常用的古菌基因敲除技术基于尿嘧啶营养缺陷型(∆)和5-氟乳清酸抗性(5-FOA)的正负筛选标记,实现精准的基因修饰。但现有的遗传操作系统仍存在阳性克隆率低等问题,故本文基于现有系统,通过改进用于同源重组的DNA分子形式,以敲除嗜盐古菌的2基因为例,建立了一种基于线性DNA片段重组的嗜盐古菌高效基因敲除系统,该技术可以促进的基因组改造。
1 材料和方法
1.1 材料
嗜盐古菌菌株H26 (∆)和基于pBluescript II质粒骨架的敲除型质粒pTA131(2+)由英国诺丁汉大学Thorsten Allers教授提供[18],本实验室保存。
1.2 方法
1.2.1 线性DNA片段重组法基因敲除原理
首先构建敲除载体,然后PCR制备线性DNA片段。传统的基因敲除法使用的敲除质粒只在筛选标记基因的任一侧携带上下游各500 bp同源臂(图1A)。线性DNA片段基因敲除法使用的敲除质粒在2筛选标记基因下游位置插入了目标基因上下游500 bp融合同源臂,同时在上游位置插入一段目标(target)基因3¢端500 bp序列,载体构建结果如图1B所示。PCR扩增包含了目标基因3¢端500 bp序列、基因、上下游500 bp同源臂,共计3段DNA序列的整段线性DNA片段。该线性DNA片段转化菌株后,由于只有在目标基因3¢端500 bp序列与下游同源片段间同时发生同源重组,标记基因和上游片段才能够同时被整合到基因组上。其中标记基因在发生同源重组后,能够在无尿嘧啶的平板上形成克隆;在目标基因下游引入的一段上游同源片段是唯一的一段可以发生二次重组的DNA同源序列,从而限定二次重组的发生部位只能在两段上游同源片段间,发生二次重组的重组子理论上都敲除了目标基因,如图1C所示。

图1 基于线性DNA同源片段的基因敲除原理
A:传统基因敲除法的环状载体模式图;B:线性DNA片段基因敲除法的敲除载体模式图;C:线性DNA片段基因敲除原理图。
1.2.2基因敲除载体构建
用细菌基因组DNA小量提取试剂盒(购自天根生物科技有限公司),提取H26基因组DNA,用质粒小量提取试剂盒(购自捷瑞生物科技有限公司)提取pTA131质粒载体,1%琼脂糖凝胶电泳鉴定基因组和pTA131质粒质量和浓度。利用无缝克隆试剂盒(购自上海远见生物科技有限公司)构建敲除载体。采用重叠PCR技术获得插入载体的上下游融合片段:即先用特异性引物扩增目的基因上、下游各自500 bp同源序列,再用两侧引物进行重叠PCR得到融合同源片段。利用克隆位点两侧的一对引物,通过PCR将pTA131载体线性化。线性化载体和待克隆DNA片段混合后,经重组酶处理5~30 min,转化DH5α感受态细胞,涂布于含100 μg/mL氨苄的LB平板。挑取单克隆于液体LB中培养,取1 μL菌液进行菌落PCR,鉴定阳性克隆。
插入了上下游同源片段的环状质粒可以直接用于传统基因敲除的一次重组。而用于线性化DNA片段重组法的敲除质粒在插入上下游同源片段后,需要继续在标记基因的另一侧插入目的基因3′端长500 bp的DNA片段。将构建好的敲除质粒作为模板,利用表1中引物4和9通过PCR扩增线性DNA同源片段,进行一次重组。载体构建过程中所用的KOD-plus DNA聚合酶购自Toyobo公司,寡核苷酸引物(由Invitrogen公司合成)序列见表1。
1.2.3DNA转化
具体操作方法参照文献[14]。在Hv-YPC培养基中,45℃培养10 mL的H26至650=0.8,25℃下,6000 r/min离心8 min收集菌体;用2 mL缓冲型原生质体化溶液(Buffered spheroplasting solution)轻柔地悬浮菌体,再次离心收集菌体;然后再次用600 μL的缓冲型原生质体化溶液轻柔地悬浮菌体,将其分成3管,分别加入0.5 mol/L EDTA(pH 8.0) 20 μL,翻转混匀,在室温下放置10 min,让其形成球状体。同时准备30 μL的DNA样本溶液(10 μL DNA水溶液(线状或环状质粒DNA 各1~2 μg,ddH2O作为阴性对照)、15 μL非缓冲型原生质体化溶液(Unbuffered spheroplasting solution)、0.5 mol/L EDTA(pH 8.0)5 μL)和800 μL 60% PEG 600溶液(480 μL PEG600,320 μL非缓冲型原生质体化溶液)。将DNA样本加入到球状体细胞溶液,取250 μL PEG600溶液,加入球状体细胞,温柔水平旋转10 min,在室温下放置30 min。然后加入1.5 mL原生质体稀释液(spheroplast dilution solution),旋转混匀后在室温下放置2 min,25℃下,6000 r/min离心8 min弃上清;加入1 mL菌体活化溶液(regeneration solution),不需要混匀;45℃静置1.5~2 h后混匀,再在45℃旋转摇动3~4 h。25℃下,6000 r/min离心8 min收集菌体,用转化稀释液(Transformant dilution solution)轻柔悬浮菌体,清洗2次,去除细胞中的尿嘧啶,最后用1 mL的转化稀释液轻柔重悬菌体;取100 μL稀释10-0、10-1、10-2、10-3倍后涂布Hv-Ca平板,45℃培养5 d。

表1 构建H. volcanii xpd2敲除质粒的引物序列
注:酶切位点用下划线标示,酶切位点依次为HⅠ(GGATCC)、RⅠ(GAATTC)、Ⅰ(CATATG)。
1.2.4基因敲除株鉴定
从一次重组平板上挑取3个单克隆于5 mL Hv-YPC培养基培养,用菌落PCR进一步验证一次重组阳性克隆,将一次重组阳性克隆在Hv-YPC培养基中以1:500稀释倍数传代2~3次。将传代后新鲜菌液(100 μL)稀释10-2、10-3、10-4倍,涂布于含有50 mg/mL 5-氟乳清酸(5-FOA)和10 mg/mL 尿嘧啶(uracil)的Hv-Ca二次筛选平板。挑取20个二次筛选平板上的单克隆于5 mL Hv-YPC培养基培养,菌落PCR检验基因是否被成功敲除。取1 μL菌液于99 μL ddH2O中稀释,取1 μL稀释液作为PCR模板。PCR扩增体系为50 μL,包括:1 μL稀释后菌液,0.3 μmol/L引物,200 μmol/L dNTPs,2 mmol/L MgCl2,1×PCR缓冲液,5% DMSO,2.5 U rDNA 聚合酶(购自TaKaRa公司)。PCR扩增条件为:95℃10 min;95℃30 s,60℃30 s,72℃3 min,30个循环;最后再72℃延伸5 min。
1.2.5基因敲除株表型分析
将成功删除基因的敲除株接种于5 mL的Hv-YPC液体培养基中,45℃过夜培养至650≈0.4;用18%盐水培养基将菌液梯度稀释10–1~10–7倍,取20 μL菌液置于Hv-YPC平板表面,让其自然晾干(大约20 min左右);将接种的Hv-YPC平板(打开平板盖)在UV (254 nmol/L,1 J/m2/s)灯下,照射不同时间(0 s、30 s、60 s、90 s、120 s、180 s、240 s);将Hv-YPC平板置于黑色避光塑料袋,于45℃培养5~7 d。
2 结果与分析
2.1 Xpd2基因敲除质粒的构建结果
载体具体构建过程详见材料方法部分,结果见图2。首先将上下游融合DNA片段插入pTA131质粒(图2A)。首先成功扩增出了基因两侧的上、下游同源DNA片段(图2A(a),泳道1和2),长度都是500 bp。通过重叠PCR,得到上、下游同源片段的融合DNA片段,长度1 kb(图2A(a),泳道3)。为了进行无缝克隆,在融合片段的两侧添加了15 bp的同源DNA序列,用于与线性化pTA131载体(图2A(a)泳道4)杂交配对,形成重组DNA。上下游融合片段和线性化载体经体外DNA重组反应转化DH5α,菌落PCR表明阳性克隆率达70%左右(图2A(b))。插入上下游融合同源片段的载体命名为pTA131- UDxpd(可用于传统基因敲除)。同理,在pTA131- UDxpd的基础上再插入目标基因编码区3¢端的500 bp序列(图2B)。目标基因3¢端500 bp片段可以成功扩增(图2B(a)泳道1),并在末端引入了用于无缝克隆的15 bp同源序列(图2B(a)泳道2)。目的基因3¢端500 bp片段与PCR线性化的pTA131-UDxpd载体无缝克隆的阳性克隆率为100%(图2B(b))。最后,将构建好的敲除载体作为模板,用下游同源片段与目标基因3¢端片段的外侧引物,在6个复性温度梯度下PCR扩增用于基因敲除的线性化DNA片段。6个复性温度(53、54.5、57、、60、62、64℃)下3.0 kb的目的条带均能够有效扩增,但存在一条短的非目标带(图2C)。由于杂带长度只有1 kb左右,重组不会将基因整合到染色体上,因此,PCR产物纯化后直接转化H26感受态细胞。线性DNA重组片段含有3段DNA功能区(图1B),与染色体同源重组,使标记基因和上游片段同时被整合到基因组上,菌株获得基因型。

图2 H. volcanii xpd2基因敲除质粒构建
A:基因上下游融合同源片段的克隆。(a):基因上下游500 bp同源序列和pTA131载体PCR结果。扩增引物对依次为1和2(上游同源片段,泳道1)、3和4(下游同源片段,泳道2)、通用引物5和6(上、下游同源片段,泳道3),7和8(线性化载体片段,泳道4)。(b):基因上下游融合同源片段的基因克隆菌落PCR鉴定结果。B:基因3¢端500 bp片段的克隆。(a):基因3¢端500 bp片段和pTA131-UDxpd载体的PCR扩增结果。泳道1:基因3′端500 bp片段;泳道2:两端附加了用于无缝克隆的15 bp同源臂的基因的3¢端500 bp片段;泳道3:pTA131-UDxpd载体PCR线性化片段。(b):基因3¢端500 bp片段的克隆菌落PCR鉴定结果。C:用于基因敲除的线性化DNA片段制备。通过PCR(引物对9/4)制备用于转化的线性化DNA片段,用于一次同源重组。泳道1~6是不同复性温度(53℃、54.5℃、57℃、60℃、62℃、64℃)下的PCR扩增结果。
2.2 Xpd2基因敲除重组子的筛选
2.2.1 线性DNA片段基因敲除法一次重组筛选结果
5~7 d后,在一次重组Hv-Ca筛选平板长出克隆,如图3A。因为线性DNA片段重组法敲除时,DNA重组反应必须同时发生在两个同源片段之间,标记基因才能整合到基因组,Hv-Ca平板才能长出克隆,所以一次重组平板长出的克隆数目不多,与传统的同源重组法相比,克隆数目大大减少。随机挑取3个克隆,用引物1和4进行一次重组菌落PCR鉴定,结果见图3B。一次重组PCR结果显示有两条带,一大一小。从图1C线性DNA片段重组的原理图中可以看出,发生一次重组时线性同源片段和基因组同源重组后整合在一起,存在两段上游同源臂和一段下游同源臂,由于上游引物有两处配对区,所以PCR时会出现两条带:小条带只由上下游同源片段构成,长约1 kb,产量高;大条带包括待敲除序列、标记基因、插入的上游500 bp同源序列,长度接近3 kb,产量低。
2.2.2 二次重组筛选结果
由于基于线性DNA片段的一次重组使得目标基因下游插入了一段上游同源序列,使得二次重组的发生部位只能在两段上游同源片段间进行。因此,发生二次重组的重组子理论上都敲除了目标基因和标记基因,都是阳性克隆。图4A是基因二次重组平板上的菌落生长情况;图4B是线性DNA 片段重组法20个二次重组克隆的菌落PCR鉴定结果,阳性克隆率约为65%左右。图C为利用环状质粒进行传统基因敲除的菌落PCR结果,其中扩增成功了7个克隆的菌落PCR,鉴定到2个克隆删除了目标基因,为阳性克隆,阳性克隆率为29%左右。从这一结果看出,基于线性DNA片段重组法敲除目的基因的成功率明显增强。
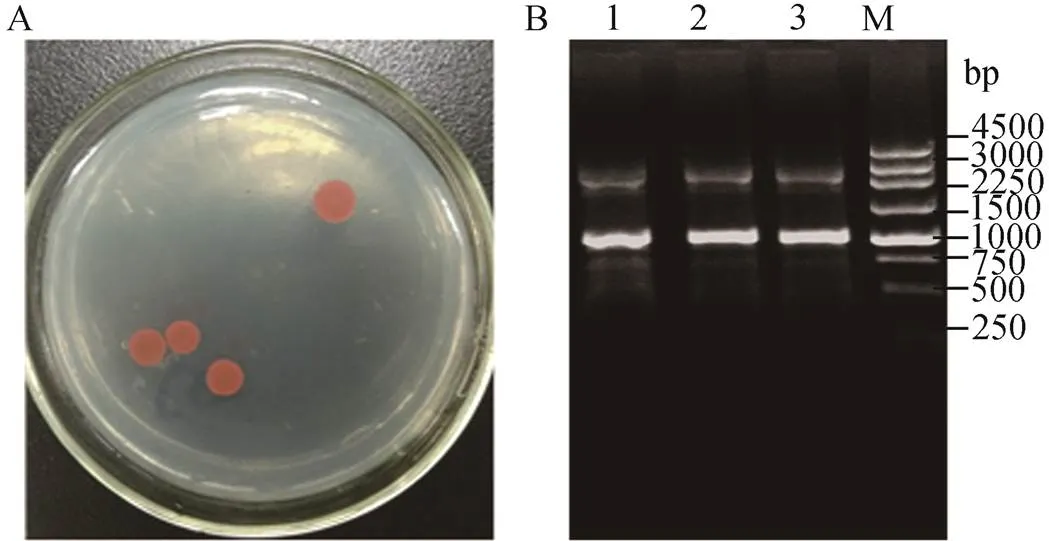
图3 xpd2基因敲除一次重组筛选结果
A:线性DNA片段基因敲除的一次重组形成的重组子平板生长情况;B:一次重组菌落PCR鉴定结果。泳道1~3代表3个独立的一次重组子;M:DNA marker。
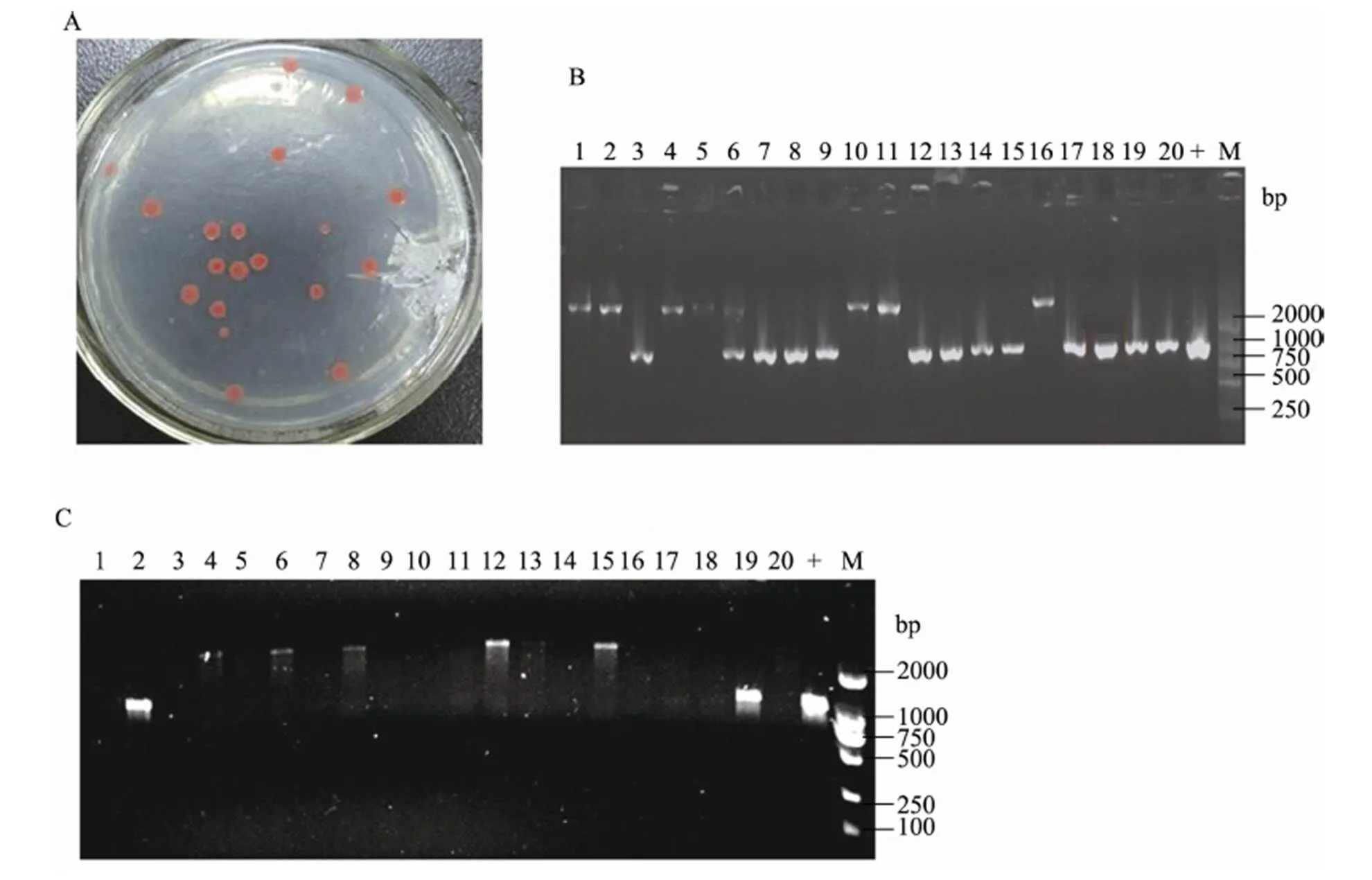
图4 xpd2基因敲除二次重组筛选结果
A:二次重组子的平板生长情况;B:线性DNA片段同源重组法敲除基因的二次重组PCR鉴定结果(M:DNA marker;1~20为20个独立的二次重组子的菌落PCR);C:传统同源重组法敲除基因的二次重组PCR鉴定结果(M:DNA marker;1~20为20个独立的二次重组子菌落PCR)。
2.3 Xpd2基因敲除后的表型研究
基因位于主染色体的37234~ 39438区域,编码一种DNA解螺旋酶,可能参与核苷酸切除修复和RNA转录过程[19~23]。初步实验结果表明:删除后,突变株的生长表型与野生株没有明显变化。由于嗜盐菌菌株中有两个()基因,分别为(, HVO_1351)和,HVO_0039),功能上可能存在冗余效应。下一步研究将在此基础上继续敲除基因,获得基因完全缺失型菌株,进而从生长表型等方面全面深入探究基因的功能。
3 讨 论
目前常用的基因敲除方法都是基于环状自杀质粒的传统同源重组法,由于在二次重组时存在回复到野生型菌株的可能,导致二次重组子中敲除目的基因的阳性突变菌株比例很低,有时需要挑取几十个克隆,才能筛选到阳性克隆,甚至筛选不到阳性克隆。本文建立了基于线性DNA片段的基因敲除技术,该方法一次重组时在目标基因的下游引入一段上游同源片段和标记,从而限定二次重组的发生部位只能发生在两段上游同源片段间,使得发生二次重组的重组子理论上都敲除了目标基因,理论上的阳性克隆率达到100%。采用本文的线性DNA片段基因敲除方法,基因的敲除效率达到65%以上。目的基因敲除效率没有达到100%的可能原因是基因自发突变或者污染了缺失型嗜盐菌,这些突变或缺失的菌株都可以在5-FOA平板上生长,但它们的2基因并未敲除。当然,也有可能是单一基因敲除实验的误差所致。总之,该方法比基于环状质粒的经典基因敲除方法,敲除效率提高了2.2倍(图4)。由此可见,线性DNA片段重组法显著提高了基因敲除突变体的阳性克隆率。由于嗜盐菌H26生长缓慢,需要5~7 d才能够形成克隆,有效的基因敲除及鉴定技术对快速构建突变株至关重要。本文综合运用线性DNA片段重组法和简单的菌落PCR鉴定技术,为快速获得嗜盐菌的基因敲除突变株提供了一种有效方法,对其他嗜盐菌遗传操作系统也具有一定借鉴作用。
[1] Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya.,1990, 87(12): 4576–4579.
[2] Allers T, Mevarech M. Archaeal genetics—the third way.,2005, 6(1): 58–73.
[3] Kelman Z, White MF. Archaeal DNA replication and repair., 2005, 8(6): 669–676.
[4] Wu Z, Liu J, Yang H, Xiang H. DNA replication origins in archaea., 2014, 5: 179.
[5] Norais C, Hawkins M, Hartman AL, Eisen JA, Myllykallio H,Allers T. Genetic and physical mapping of DNA replication origins in., 2007, 3(5): e77.
[6] Allers T, Ngo HP. Genetic analysis of homologous recombination in Archaea:as a model organism., 2003, 31: 706–710.
[7] Liu HL, Han J, Liu XQ, Zhou J, Xiang H. Development of-based gene knockout systems for genome-wide manipulation of the archaeaand., 2011, 38(6): 261–269.
[8] Liu HL, Wu ZF, Li M, Zhang F, Zheng HJ, Han J, Liu JF, Zhou J, Wang SY, Xiang H. Complete genome sequence of, a Model Haloarchaeon for studying genetics, metabolism, and virus-host interaction., 2011, 193(21): 6086–6087.
[9] Wagner M, van Wolferen M, Wagner A, Lassak K, Meyer BH, Reimann J, Albers SV. Versatile genetic tool box for the crenarchaeote sulfolobus acidocaldarius., 2012, 3: 214.
[10] Zhang CY, Guo L, Deng L, Wu YX, Liang YX, Huang L, She QX. Revealing the essentiality of multiple archaeal pcna genes using a mutant propagation assay based on an improved knockout method., 2010, 156(11): 3386–3397.
[11] Bridger SL, Clarkson SM, Stirrett K, DeBarry MB, Lipscomb GL, Schut GJ, Westpheling J, Scott RA, Adams MWW. Deletion strains reveal metabolic roles for key elemental sulfur-responsive proteins in., 2011, 193(23): 6498–6504.
[12] Sato T, Fukui T, Atomi H, Imanaka T. Targeted gene disruption by homologous recombination in the hyperthermophilic archaeonKOD1., 2003, 185(1): 210–220.
[13] Hartman AL, Norais C, Badger JH, Delmas S, Haldenby S, Madupu R, Robinson J, Khouri H, Ren QH, Lowe TM, Maupin-Furlow J, Pohlschroder M, Daniels C, Pfeiffer F, Allers T, Eisen JA. The complete genome sequence ofDS2, a model archaeon., 2010, 5(3): e9605.
[14] Cline SW, Lam WL, Charlebois RL, Schalkwyk LC, Doolittle WF. Transformation methods for halophilic archaebacteria., 1989, 35(1): 148–152.
[15] Allers T, Ngo HP, Mevarech M, Lloyd RG. Development of additional selectable markers for the halophilic archaeonbased on theandgenes., 2004, 70(2): 943–953.
[16] Bitan-Banin G, Ortenberg R, Mevarech M. Development of a gene knockout system for the halophilic archaeonby use of thegene., 2003, 185(3): 772–778.
[17] Holmes M, Pfeifer F, Dyall-Smith M. Improved shuttle vectors forincluding a dual-resistance plasmid., 1994, 146(1): 117–121.
[18] Allers T, Barak S, Liddell S, Wardell K, Mevarech M. Improved strains and plasmid vectors for conditional overexpression of his-tagged proteins in., 2010, 76(6): 1759–1769.
[19] Liu HT, Rudolf J, Johnson KA, McMahon SA, Oke M, Carter L, McRobbie AM, Brown SE, Naismith JH, White MF. Structure of the DNA repair helicase XPD., 2008, 133(5): 801–812.
[20] Lehmann AR. The xeroderma pigmentosum group D () gene: one gene, two functions, three diseases., 2001, 15(1): 15–23.
[21] Winkler GS, Araújo SJ, Fiedler U, Vermeulen W, Coin F, Egly JM, Hoeijmakers JHJ, Wood RD, Timmers HTM, Weeda G. TFIIH with inactive XPD helicase functions in transcription initiation but is defective in DNA repair., 2000, 275(6): 4258–4266.
[22] Kou HP, Zhou Y, Charlotte Gorospe RM, Wang ZG. Mms19 protein functions in nucleotide excision repair by sustaining an adequate cellular concentration of the TFIIH component Rad3., 2008, 105(41): 15714–15719.
[23] Woods WG, Dyall-Smith ML. Construction and analysis of a recombination-deficient () mutant of., 1997, 23(4): 791–797.
(责任编委: 何群)
An efficient genetic knockout system based on linear DNA fragment homologous recombination for halophilic archaea
Xiaoli Wang, Chuang Jiang, Jianhua Liu, Xipeng Liu
With the development of functional genomics, gene-knockout is becoming an important tool to elucidate gene functions. As a good model strain for archaeal genetics,has received more attention. Although several genetic manipulation systems have been developed for some halophilic archaea, it is time-consuming because of the low percentage of positive clones during the second-recombination selection. These classical gene knockout methods are based on DNA recombination between the genomic homologous sequence and the circular suicide plasmid, which carries aselection marker and two DNA fragments homologous to the upstream and downstream fragments of the target gene. Many wild-type clones are obtained through a reverse recombination between the plasmid and genome in the classic gene knockout method. Therefore, it is necessary to develop an efficient gene knockout system to increase the positive clone percentage. Here we report an improved gene knockout method using a linear DNA cassette consisting of upstream and downstream homologous fragments, and themarker. Gene deletions were subsequently detected by colony PCR analysis. We determined the efficiency of our knockout method by deleting thegene from thegenome, with the percentage of positive clones higher than 50%. Our method provides an efficient gene knockout strategy for halophilic archaea.
halophilic archaea; gene knockout; homologous recombination; linear DNA recombination
2014-10-24;
2015-01-12
国家自然科学基金项目(编号31371260)资助
王小利,硕士,专业方向:DNA修复。E-mail: 650914@sjtu.edu.cn
刘喜朋,博士,副教授,研究方向:微生物遗传学。E-mail: xpliu@sjtu.edu.cn
10.16288/j.yczz.14-366
2015-3-11 9:17:12
http://www.cnki.net/kcms/detail/11.1913.R.20150311.0917.001.html
