基于高通量测序的宏基因组学技术在动物胃肠道微生物方面的研究进展
2015-02-02张莺莺昝林森
吴 森, 张莺莺, 昝林森,2*
1.西北农林科技大学动物科技学院, 陕西 杨凌 712100;2.西北农林科技大学国家肉牛改良中心, 陕西 杨凌 712100
植物是自然界中普遍存在的重要可再生资源,且存量巨大,但大多数是人类不能直接利用的。草食性动物及杂食性动物具有其他动物所没有的能力——消化植物纤维,将植物纤维转化为肉、蛋、奶等产品供人类利用。这种转化作用主要为草食性动物胃及肠道寄生微生物对植物细胞壁中复杂碳水化合物的降解作用,包括对木聚糖、果胶、纤维素等的降解作用。揭示动物胃肠道中主要微生物的作用机理,对人工干预和提高植物源性资源向动物产品转化的效率,促进动物科学精准饲养,以及植物源性材料的人工发酵等具有重要意义。
随着技术的发展,基于高通量测序技术的宏基因组技术,通过提取特定环境中的微生物总DNA、构建文库,可以获得大量微生物群落种类、丰度及相关生物学信息。相对于传统微生物研究方法的分离、培养,更能获得大量的不可培养的微生物信息,是目前研究特定环境微生物的重要技术手段。本文综述了常见Roche 454、Illumina基因分析和ABI SOLiD测序三大高通量测序平台的宏基因组技术在动物胃肠道微生物的研究应用进展,旨在为动物科学生产及微生物发酵等相关领域的研究工作提供科学指导。
1 基于高通量测序的宏基因学技术在反刍动物胃部微生物方面的应用研究
大多数哺乳动物缺乏消化植物细胞壁(纤维素、半纤维素、果胶、β-葡聚糖、低聚糖和木质素等)的酶,而反刍动物等草食动物可通过瘤胃降解消化植物,利用在瘤胃中微生物的厌氧发酵产生的短链脂肪酸,以获得所需的大部分营养,约是动物所需营养的60%~80%,其余20%左右的能量在大肠部位消化吸收[1]。由于胃肠道微生物种类繁多、丰度不一、作用多样,常规分离提取培养等方法难以全面了解这些微生物发酵的分子机理。随着现代DNA、RNA测序技术和生物学信息分析技术的发展,现代宏基因组学为人们研究反刍动物胃肠道微生物群落结构功能提供了新的思路与方法。
1.1 甲烷排放相关研究
牛、羊等反刍动物的瘤胃是一个巨大的微生物资源库,拥有丰富的菌种。主要有细菌、古菌、原虫和真菌,是迄今为止已知的分解纤维物质能力最强的天然发酵罐,可在厌氧条件下将纤维、果胶、淀粉和蛋白质等降解为短链脂肪酸等营养物质供机体吸收。瘤胃中的真菌对发酵作用影响显著,相对于纤维素,其能更有效地降解木质素[2]。反刍动物瘤胃中含量较少的古菌主要是甲烷菌属,且甲烷产量可以作为衡量瘤胃对日粮利用效率的重要指标[3]。但是,甲烷气体是引起温室效应的主要因素之一,畜牧业中的甲烷排放对引起全球变暖的温室效应有着不可推卸的责任。畜牧生产中甲烷气体主要是植物在反刍动物瘤胃的厌氧环境下由甲烷菌属的相关产甲烷菌发酵后产生的[4]。由于产甲烷菌群落组成复杂,难以完全鉴定,特别是在动物体内环境中[5]。传统检测反刍动物胃肠的甲烷菌是通过针对已知的甲基辅酶M还原酶a亚基(methyl coenzyme-M reductase A,mcrA)或SSU(small-subunit)rRNA数据库设计引物,qPCR扩增检测特定类群微生物数量;或者用一些化学标记物检测产甲烷菌代谢附属物,来分析产甲烷古菌的代谢情况[6]。但因为数据库更新慢、引物特异性低和代谢附属物易受体内其他条件影响等因素,造成检测精度低、获得的信息量少。例如,Tymensen[7]用5种常用SSU rRNA引物扩增检测黑安格斯牛瘤胃液中的古菌,结果之间有一些偏差。但高通量测序技术以其高输出量和高解析度的优点,可更加全面的检测反刍动物瘤胃复杂微生物环境中古菌的生物学信息。Snelling等[8]运用传统Sanger 16S rRNA基因扩增及Illumina平台高通量16S rRNA基因测序,分析了苏格兰高地绵羊瘤胃中产甲烷的甲烷短杆菌、甲烷杆菌及甲烷球菌,对比测序结果分析后发现Illumina平台高通量的V6-V8 16S rRNA基因测序能更高效用于检测瘤胃古菌群落的研究。
1.2 植物降解相关研究
瘤胃对植物的降解作用,主要是瘤胃中原虫、真菌及细菌微生物发酵产生可降解植物的相关酶类。传统研究反刍动物瘤胃微生物对植物降解作用的方法,需要对瘤胃内容物提取、分离后培养鉴定,方法繁琐,获得数据量少,且有大量不可培养的微生物流失。运用高通量测序的宏基因组技术可获得大量与植物降解相关的微生物信息。例如,Singh等[9]对印度水牛瘤胃中的微生物做高通量测序后,获得137 270个contigs(contiguous sequences)。鉴定后有2 614个具有编码降解酶功能的contigs,包括聚糖水解酶类(glycoside hydrolases,GH: 1943 contigs)、糖结合模块(carbohydrate binding module,CBM: 23 contigs)、糖基转移酶(glycosyl transferase,GT: 373 contigs)、糖类酯酶(carbohydrate esterases,CE: 259 contigs)及多聚糖裂解酶类(polysaccharide lyases,PE: 16 contigs);Rosewarne等[10]对饲喂无芒虎尾草的印度阉牛采用瘤胃瘘管法采集瘤胃食糜后,以Roche 454平台16S rRNA基因高通量测序,对瘤胃微生物生物信息分析发现,有50多个显著的多聚糖利用作用位点(sus-like polysaccharide utilization loci,PULs),且这些位点在草食动物中广泛存在,主要起降解植物的作用;Wang等[11]通过高通量测序对瘤胃木质纤维素降解相关细菌检测后,鉴定出了大量木质纤维素特异降解性细菌;Sarubbi等[12]采用基于高通量测序的宏基因组技术分析了奶牛瘤胃微生物对高粱、玉米青贮的降解作用,对关键性纤维素分解微生物及木聚糖分解相关微生物的丰度、组成测序后,关联日增重与产奶量后发现瘤胃对两者的降解能力相同,无显著差异,从而得知高粱、玉米青贮可互替代。应用高通量测序的宏基因组技术能够方便的获得大量植物降解相关微生物的生物学信息,为动物营养学等提供参考。
1.3 与抗病致病基因筛选相关的研究
动物体内微生物与动物健康、人类食品安全密切相关。尽管大部分牛瘤胃肠道微生物都已经鉴定出来,但是少有整个群落基因的功能性研究,特别是关于抗病、致病性微生物的研究。基于高通量测序技术的宏基因组学研究,在单方面获得微生物多样性信息的同时,可以从整个微生物环境的层面来获得与抗病、致病相关的微生物信息。如2012年,Singh等[13]对印度水牛瘤胃内容物进行了高通量宏基因组测序分析,在获得瘤胃微生物多样性信息的同时,研究其抗菌素耐药性及细菌毒力时发现,测序结果中有6.44%的基因序列为病毒相关基因,与抗生素抵抗和毒素生成有关,且这一数据高于肉鸡盲肠微生物的5.39%和奶牛瘤胃微生物的4.43%;为研究瘤胃微生物与细菌及抗菌素抗性之间的关系,Reddy等[14]对饲喂青、干粗饲料的印度水牛瘤胃液及食糜中的微生物应用高通量测序宏基因组技术,发现在门水平,所有组中拟杆菌门和厚壁菌门分别占各组微生物的第一和第二,且瘤胃液、固体食糜中,关于氧化性应激应答和抗病性功能基因序列基本相同,但抗氟喹诺酮、多耐药性外排泵(multiple drug resistance(MDR) efflux pumps)和抗甲氧西林相关基因的表达却交叉分布于11、9、14种细菌中。另外,细菌与噬菌体应答、包装及噬菌体原体有关,链球菌噬菌体与拟杆菌门、厚壁菌门和变形菌门细菌分布有关。高通量测序的宏基因组技术不仅可以对反刍动物瘤胃微生物做出基础的菌群群落分析,也可相对定量的做出定性分析。
1.4 日粮组成对反刍动物瘤胃微生物的影响
瘤胃微生物稳态受多方面的影响,其中不同日粮组成对瘤胃微生物影响显著。在日粮组成对反刍动物瘤胃微生物影响的相关研究方面,采用基于高通量测序的宏基因组技术,对不同日粮处理的反刍动物瘤胃样检测后,可获得远多于传统方法的大量微生物的生物学信息,便于更准确的分析日粮与瘤胃微生物的关系。如Pitta等[15]用不同精粗比例的饲料饲喂印度水牛6周后,采集瘤胃食糜、瘤胃液后进行高通量测序共获得333 851 pyrotags。做系统进化分析(phylogenetic analysis)后显示,不同比例粗饲料日粮(50%、75%、100%粗饲料)可引起瘤胃微生物的显著性差异;不同日粮成分可引起微生物群落组成的不同。在属水平,日粮的变化会引起微生物群落组成的显著差异:在100%粗饲料组中,瘤胃球菌属(Ruminococcus)和纤维杆菌属(Cellulomonas)含量与其他两组相比显著增高;在50%粗饲料组中普氏菌属(Prevotella)含量显著高于其他组。Patel等[16]以不同青草、干草粗饲料与不同精饲料比例的日粮饲喂水牛,提取瘤胃样本进行高通量宏基因组测序,分析碳水化合物激活酶(carbohydrate active enzymes,CAZymes),共获得2 597个被CAZyme analysis toolkit (CAT)识别的CAT重叠区(contigs encoding);饲喂青草粗饲料的水牛瘤胃中多糖降解细菌属(如纤维杆菌属、普氏菌属、拟杆菌属、放线菌属和瘤胃球菌属细菌)丰度显著高于饲喂干草粗饲料组,且果胶消化酶类(如果胶裂合酶1、胶裂合酶10及多糖降解酶28)显著高于饲喂干草粗饲料组。相似的,Parmar等[17]对8头以青草、干草为粗饲料的水牛瘤胃微生物做高通量宏基因组测序分析后发现,两种粗饲料处理水牛瘤胃微生物均是:在门水平上,拟杆菌占主要地位;在属水平上,普氏菌属占最多数。相对于瘤胃液,固体食糜中厚壁菌门与拟杆菌门微生物比率较高。以干草为粗饲料的牛瘤胃固体食糜中,拟杆菌门数量随着粗饲料的增加显著升高。且在属水平,随着粗饲料比例的增加,梭菌(Clostridium)含量显著增加。另外,对饲喂青草、干草组的瘤胃液中糖苷水解酶类及碳水化合物激活酶类(cellulosome functional)基因表达量检测发现,随着日粮中纤维素含量的增加,糖苷水解酶3(GH3)水平升高。Roggenbuck等[18]对饲喂多叶苜蓿和非洲当地热带牧草的长颈鹿瘤胃液及固体内容物进行16S rRNA基因扩增及Roche 454平台高通量测序后发现,瘤胃液与内容物微生物之间没有显著差异;不同饲草饲喂的长颈鹿瘤胃微生物中厚壁菌门及拟杆菌门细菌均表现出较高丰度,其中普氏菌属和甲烷短杆菌属(Methanobrevibacter)丰度最高;而且在测序结果中约20%的细菌序列为首次发现的细菌序列。这些均是调整日粮后,应用高通量测序技术所得到的丰富详细的宏基因组学信息,极大的方便了学者对日粮与动物胃部微生物关系的把握。
1.5 牛不同发育阶段瘤胃微生物的变化研究
反刍动物瘤胃微生物对机体的生理活动非常重要。但是,在以牛为典型代表的反刍动物的生长过程中,微生物群落的种类、丰度如何变化呢?Jami等[19]选取了从出生1 d到2岁的成年以色列荷斯坦奶牛,研究宿主年龄变化对瘤胃微生物群落的影响。其中,6月龄和2岁龄采用70%精料+30%粗饲料饲喂,2月龄断奶牛饲喂母乳和特定的固体补充精料,初生犊牛6头仅饲喂初乳。结果发现,瘤胃发酵整个过程是一个从有氧发酵到无氧发酵的过程,微生物群落逐渐丰富活跃。其中初生犊牛瘤胃中几乎没有成年牛瘤胃中的微生物;6月龄与2岁牛虽然饲喂饲料相同,但瘤胃微生物菌群有着显著差异;相对于初生牛,成年牛瘤胃微生物环境更加均衡、严格,菌群更趋于多样化,2岁时瘤胃微生物群落趋于成熟稳定;从出生到成年,牛瘤胃中微生物在门水平主要由变形菌门、厚壁菌门和拟杆菌门的细菌组成,占主要地位,且随着年龄的增长变形菌门细菌的数量下降,2月龄后拟杆菌门数量虽有变化但基本保持一定水平(图1);在出生3 d后普氏菌属、粪球菌属(Coprococcus)和水解纤维素有关的瘤胃球菌属等细菌逐渐增多并表现出一定丰度,逐渐完善了瘤胃功能(表1)。这一趋势与一项研究人类婴儿随着年龄增长大肠微生物变化的报道类似:随着机体的成熟,消化机能逐步完善,体内微生物种群会逐渐丰富[20]。Koenig等[21]关于婴儿肠道微生物的研究印证了这一点,同时也指出儿童肠道微生物群落在2.5岁后基本趋于稳定。高通量测序的宏基因组技术为研究各个发育阶段微生物提供了充足的数据支持,使得整个发育阶段微生物变化脉络更加清晰完备,且研究更加简单易行。
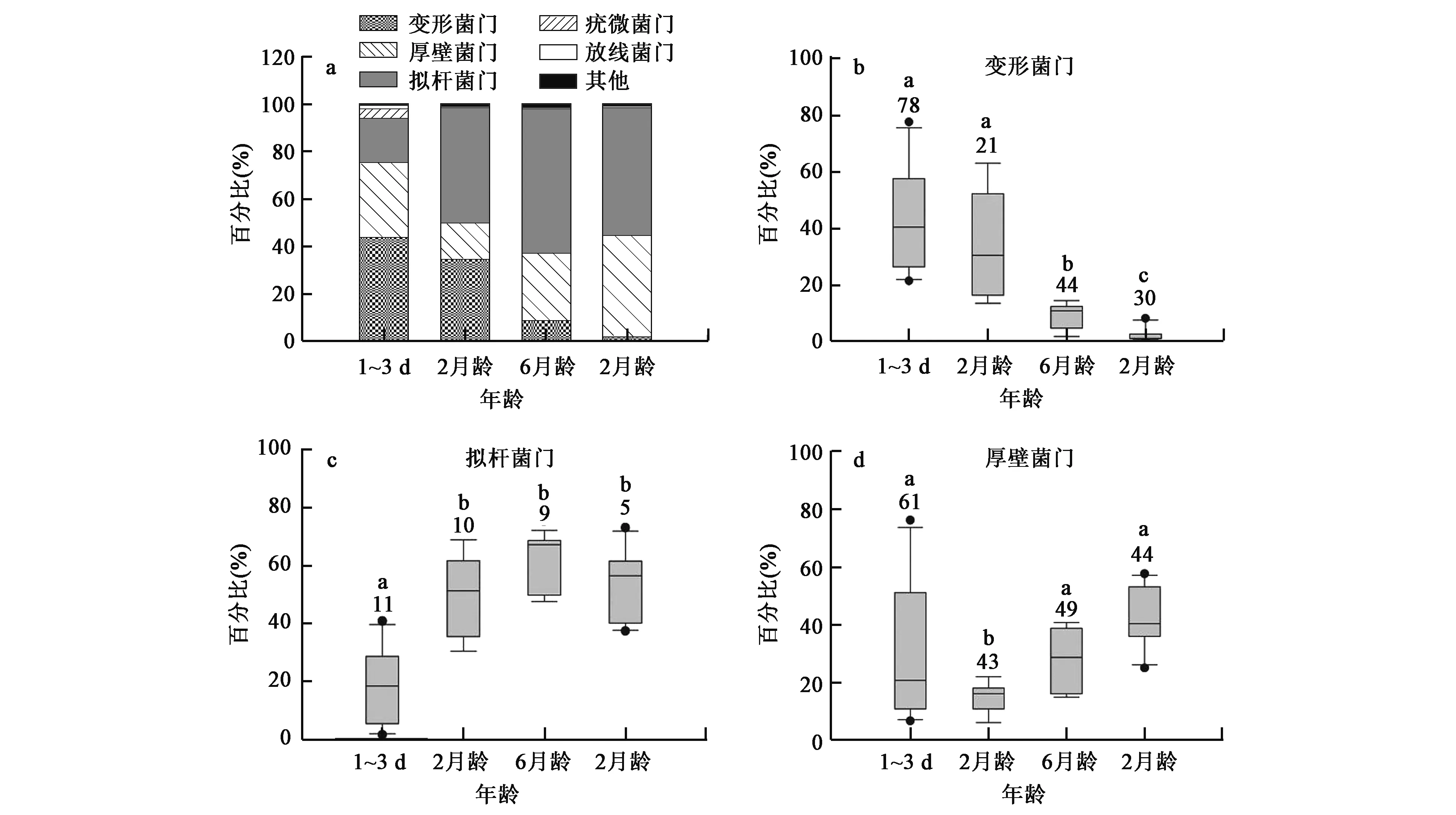
图1 不同年龄阶段牛瘤胃微生物门水平变化[19]Fig.1 Microbial composition in different age stages of bovine rumen at phylum level[19].注:a:各年龄阶段牛瘤胃微生物门属水平分布; b、c、d:不同年龄阶段各组变形菌门、拟杆菌门和厚壁菌门微生物所占比例,图中不同小写字母表示差异显著(P<0.05)。

门属种1日龄(%)3日龄(%)2月龄(%)6月龄(%)2岁龄(%)拟杆菌门Bacteroidetes普氏菌属Prevotella3.85±3.3548.88±6.9859.68±5.1556.66±5.28拟杆菌目*Bacteroidales1.48±0.820.34±0.160.52±0.140.27±0.05梭菌属Clostridium2.16±1.021.81±0.350.86±0.155.24±1.194.60±0.67毛旋菌科*Lachnospiraceae0.08±0.023.58±0.735.02±0.737.02±1.03琥珀酸菌属Succiniclasticum0.22±0.130.76±0.081.35±0.172.01±0.36粪球菌属Coprococcus0.13±0.111.24±0.381.68±0.231.88±0.20瘤胃球菌属Ruminococcus0.20±0.131.30±0.441.50±0.201.60±0.11瘤胃菌科*Ruminococcaceae0.67±0.280.80±0.185.30±1.345.56±0.82丁酸弧菌属Butyrivibrio1.41±0.862.44±0.619.17±1.66梭菌目*ClostridialesFamilyXIII.IncertaeSedis0.15±0.060.99±0.211.38±0.25厚壁菌门Firmicutes月行单孢菌属Selenomonas0.13±0.020.27±0.071.03±0.22光岗菌属Mitsuokella0.08±0.010.14±0.020.58±0.20真菌属Eubacterium0.50±0.050.56±0.070.71±0.04罗氏菌属Roseburia0.06±0.010.22±0.030.41±0.05假丁酸弧菌属Moryella0.15±0.010.10±9.03E-050.38±0.10丹毒丝菌属Bulleidia0.06±0.020.10±0.020.21±0.05毛螺旋菌属Lachnobacterium0.08±0.040.11±0.020.16±0.04颤螺旋菌属Oscillospira0.55±0.160.13±0.020.11±0.02毛螺菌属Lachnospira0.15±0.060.05±5.31E-050.08±0.02变形菌门Proteobacteria毛螺菌属Lachnospira0.15±0.060.05±5.31E-050.08±0.02琥珀酸弧菌属Succinivibrio34.45±9.478.45±2.291.41±0.32总和2.168.4495.5393.8595.23
注:各组各个年龄段微生物属水平微生物平均丰度值,未达属水平的菌目标以*。
2 基于高通量测序的宏基因组学技术在动物肠道微生物方面的应用研究
除了反刍动物瘤胃中微生物能消化植物纤维以外,在草食动物和杂食动物中,大肠是一个重要的消化器官,机体所需能量的20%左右是从大肠获取的[1]。在利用现代宏基因组技术研究大肠中微生物的消化作用方面,Ilmberger等[22]对最大的陆地植食性动物—亚洲象大肠微生物群落的研究中,选取3周龄和6岁的亚洲象粪便做 Illumina平台的高通量宏基因组检测分析后,分别获得了380个和3 000个可操作单元(operational taxonomic units,OTUs)及1.1 Gb的DNA contigs,并发现了84个GH的PULs多属于拟杆菌门,还包括一些纤维素酶基因。另外,拟杆菌门中,GH5和GH9家族酶表现出较高丰度,可能主要是在亚洲象消化纤维素过程中起作用。
在肠道微生物研究中,应用高通量测序技术可准确获得肠道微生物群落的分类、丰度信息,为人类研究肠道营养健康提供支持。Ziemer[23]取猪粪便,加纤维素、果胶木聚糖连续培养8周后,利用基于高通量测序的宏基因组技术得到575种细菌菌株,分属6门:厚壁菌门(242)、拟杆菌门(185)、变形菌门(65)、梭菌门(55)、放线菌门(23)和互养菌门(5);与核糖体数据库(ribosomal database project, RDP)比对后,约有30%的细菌为不可培养菌,有179株属于新的菌种或菌属。其中厚壁菌门主要由毛旋菌科、肠球菌科、葡萄球菌科及梭菌科I(Clostridiaceae I)组成;拟杆菌门以多形拟杆菌属、卵形拟杆菌属及B.木聚糖占主导地位;而厚壁菌和拟杆菌中的大部分菌可产生降解植物细胞壁的酶,参与植物细胞壁的降解消化,与肠道的消化功能有关。之后,Ziemer[24]还对爱荷华州立大学农场的4头经产奶牛粪便取样,分离细菌继续培养8周后同样采用高通量测序的宏基因组学技术,检测微生物后并与RDP比对发现6门、459种菌属(厚壁菌门51.9%、拟杆菌门30.9%、变形菌门11.1%、放线菌门3.5%、互养菌门1.5%、梭菌门1.1%),这也仅占所分离细菌的近98.5%;厚壁菌门主要有鞭毛菌科、瘤胃菌科、丹毒菌科及梭菌科(Clostridiaceae I);拟杆菌门主要是卵形多形拟杆菌B.及木聚糖溶解酶B.。类似地,de Oliveira等[25]对巴西Nelore阉牛瘤胃、小肠、大肠内容物做16S rRNA基因高通量测序检测后也发现厚壁菌门和拟杆菌门微生物为各个组织部位的主要菌群。Ni等[26]对24条草鱼大肠微生物做宏基因组检测后发现有116种古菌和1 112种细菌,且主要为厚壁菌门、变形菌门和梭菌门细菌。Singh等[27]采用高通量测序对饲料转化率不同的印度“MY”肉鸡粪便中微生物菌群检测后,与RDP、小分子rRNA数据库(SSU rRNA database)及SEED数据库(SEED database)比对后发现,微生物中细菌占95%以上,其次依次为真核生物(>2%)、古菌(>0.2%)和病毒(>0.2%);在门水平,两组微生物中变形菌门、厚壁菌门和拟杆菌门含量依次降低,在高、低饲料转化率肉鸡粪便样品的微生物群落中变形菌门、厚壁菌门和拟杆菌门含量依次为52.04%和78.83%、27.53%和11.97%、17.53%和7.10%。通过高通量测序的宏基因组学技术对动物肠道微生物测序,然后比对数据库,确定这些肠道微生物的主要功能,为后续动物肠道营养健康研究提供了方法。
3 基于高通量测序的宏基因组学技术在特定功能微生物筛选中的应用
草食动物可直接消化利用植物,主要是草食动物体内微生物在特定条件下可以产生相关的降解酶类,来消化降解植物细胞壁。因此,我们可以通过研究动物体内微生物降解纤维的生物机理,人工干预微生物微环境,调节微生物代谢向着人类希望的方向发展;相似地,我们也可通过对草食动物体内微生物的研究,筛选出具有特定功能的微生物应用于工业发酵等行业。因此,在应用基于高通量测序的宏基因组学技术于动物研究的同时,也可用在对特定功能微生物筛选的研究中。
例如,López-Cortés等[28]在研究牛瘤胃微生物时,发现一种新的木聚糖水解酶——乙酰基木聚糖酯酶;Palackal等[29]在牛肠道微生物中分离出复合糖基水解酶;Weimer和Stevenson[30]从牛瘤胃中分离出可产乙酸的芽孢杆菌;Liu等[31]应用宏基因组学技术从中国荷斯坦牛瘤胃中分离鉴定出一个新的厌氧型、适温、一氧化碳氧化产氢菌,并从构建的宏基因组文库中获得2个新的脂解酶RlipE1和RlipE2基因。Wang等[32]对牦牛瘤胃内容物应用高通量测序的宏基因组技术构建宏基因组文库并测序,筛选出14个属于水解酶10家族(GH10)的木聚糖酶蛋白,这些木聚糖酶的氨基酸序列相似度为20.5%~91.3%,并且7个木聚糖酶基因与木糖苷酶基因有着不同的contigs;Larsbrink等[33]对奶牛瘤胃微生物宏基因组进行高通量测序后得到27 755个碳水化合物水解酶候选基因,可编译90种候选蛋白,并且其中57%的候选基因可通过酶促作用,激活抗纤维素底物相关酶类。Pope等[34]在对驯鹿瘤胃消化木质纤维素相关微生物的宏基因组学研究中,除了发现拟杆菌门和厚壁菌门为主要作用的微生物以外,还鉴定出一种新的拟杆菌分支菌SRM-1,分析后发现其具有多个PULs,分泌20种GH和其他多种靶向碳水化合物水解酶,包括木聚糖、果胶和纤维素水解酶类。Gruninger等[35]对从饲喂青草、干草的奶牛瘤胃内容物建立宏基因组文库后鉴定出一种具有高β-葡萄糖苷酶活性的蛋白Bgxa1(beta-glucosidase/beta-xylosidase/alpha-arabinosidase gene),Bgxa1可高效抗p-硝基苯基-β-D-吡喃葡萄糖苷(p-nitrophenyl-beta-d-glucopyranoside (pNPG))、纤维二糖(cellobiose)、p-硝基苯基-β-D-木吡喃糖苷(p-nitrophenyl-beta-d-xylopyranoside (pNPX))及p-硝基苯基-α-D-阿拉伯糖苷(p-nitrophenyl-alpha-d-arabinofuranoside (pNPAf))。Bgxa1的动力学分析显示其对以上三种糖苷的催化活性高低依次为pNPG>pNPAf>pNPX;Bgxa1的催化活性是β-木糖苷酶和α-阿拉伯糖苷酶的100倍,对木质纤维素材料有较好的糖化作用。Li等[36]对饲喂中国芒草的肉牛瘤胃微生物运用宏基因组学技术,建库后筛选、克隆、鉴定出一种新的编码β-糖苷水解酶的基因unglu135B12,该基因编码一个779氨基酸的多肽链,具有一个GH3催化位点,最适酶活条件为pH 5.0、38℃。
4 展望
经过千百万年以来的自然选择和遗传进化,草食性动物和杂食性动物具有了特有的消化利用植物资源的生理功能,主要是其胃肠道适应了寄生微生物,构成了一定的微生物稳态。在这种特殊的微生物环境中,经过微生物的发酵,降解植物纤维为动物体所能吸收的营养物质,大幅提高了资源的利用率。研究并利用这一特殊的生理特性,使得人工干预微生物高效利用植物资源成为可能。而且,现今人们已经清楚的认识到生物体的生理代谢和生长发育除了受自身基因的调控外,还受到多条件的影响调控,其中就包括机体中的微生物。基于高通量测序的宏基因组技术的进步,使人们能尽可能全面地扫描到一些特殊微生物环境中的微生物基因,挖掘出更多以前尚未发现的功能性基因,以及更多的不可培养微生物,在很大程度上丰富了微生物基因库。另外,随着现代宏基因组技术在生物体内微生物研究中的应用,为人们更加全面掌握生物体机能代谢、营养调控以及疾病免疫预防等,提供了更为方便可靠的技术支持。而且,随着生物工程技术的发展,生物体内微生物的相关研究,也为生物制剂生产、生物发酵工程等微生物应用领域研究提供了帮助。
在生物体相关微生物的研究中,人类体内微生物研究走了最前面,例如在2007年及2008年,美国国立卫生研究院(NIH)及欧盟委员会(European Commission,EC)分别启动了“人类生物组计划”(Human Microbiome Project,HMP)和“人类肠道宏基因组学” (Metagenomics of Human Intestinal Tract,MetaHIT)计划,分别资助约1.15亿美元和2 770万美元,由美国、欧盟、中国、日本等多国参与合作,第一阶段资助已经完成,但由于微生物菌群的复杂特性及科技制约取得的成果与预期效果仍有差距。
虽然基于高通量测序技术的基因组技术相对于传统研究微生物的方法而言,在获得微生物数据量、可操作性等方面有了很大进步,但仍不够完善。比如,现在的高通量测序技术都是将DNA剪切为小片段后,再单个连接小片段DNA至一定的固相表面单独扩增后拼接并检测信号。DNA拼接越长难度越大,从而完美的片段阈值难以完全控制,存在一定的误差。另一方面,高通量测序越来越强的测序深度及越来越大的数据输出也导致现有算法难以完全利用如此庞大的数据量,不但造成数据的浪费并在一定程度上这也导致在后期数据分析过程中制约了高通量测序技术在宏基因组学方面的应用。但是,相信随着科技的进一步发展,下一代优化测序技术的改进,或者新研究方法的出现,一定可以使人类对于微生物研究更加深入、了解的更加透彻,从而帮助人类彻底解决生物体与微生物之间的种种谜题。
[1] Bergman E. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species[J]. Physiol. Rev., 1990,70(2): 567-590.
[2] Tuyen V D, Cone J, Baars J,etal.. Fungal strain and incubation period affect chemical composition and nutrient availability of wheat straw for rumen fermentation[J]. Bioresour. Technol., 2012,111: 336-342.
[3] Larbi A, Smith J, Kurdi I,etal.. Chemical composition, rumen degradation, and gas production characteristics of some multipurpose fodder trees and shrubs during wet and dry seasons in the humid tropics[J]. Animal Feed Sci. Technol., 1998,72(1): 81-96.
[4] Wright A D G, Klieve A V. Does the complexity of the rumen microbial ecology preclude methane mitigation?[J]. Animal Feed Sci. Technol., 2011,166: 248-253.
[5] Janssen P H, Kirs M. Structure of the archaeal community of the rumen[J]. Appl. Environ. Microbiol., 2008,74(12): 3619-3625.
[6] McCartney C A, Bull I D, Dewhurst R J. Chemical markers for rumen methanogens and methanogenesis[J]. Animal, 2013,7: 409-417.
[7] Tymensen L D, McAllister T A. Community structure analysis of methanogens associated with rumen protozoa reveals bias in universal archaeal primers[J]. Appl. Environ. Microbiol., 2012,78(11): 4051-4056.
[8] Snelling T J, Genc B, McKain N,etal.. Diversity and community composition of methanogenic archaea in the rumen of sottish upland sheep assessed by different methods[J]. PLoS ONE, 2014,9(9): e106491.
[9] Singh K M, Reddy B, Patel D,etal.. High potential source for biomass degradation enzyme discovery and environmental aspects revealed through metagenomics of Indian buffalo rumen[J]. Biomed. Res. Int., 2014, doi: 10.1155/2014/267189.
[10] Rosewarne C P, Pope P B, Cheung J L,etal.. Analysis of the bovine rumen microbiome reveals a diversity of Sus-like polysaccharide utilization loci from the bacterial phylum Bacteroidetes[J]. J. Ind. Microbiol. Biotechnol., 2014,41(3): 601-606.
[11] Wang G R, Duan Y L. Studies on lignocellulose degradation by rumen microorganism[J]. Adv. Materials Res., 2014,853: 253-259.
[12] Sarubbi F, Chiariotti A, Baculo R,etal.. Nutritive value of maize and sorghum silages: fibre fraction degradation and rumen microbial density in buffalo cows[J]. Czech J. Animal Sci., 2014,59(6): 278-287.
[13] Singh K, Jakhesara S, Koringa P,etal.. Metagenomic analysis of virulence-associated and antibiotic resistance genes of microbes in rumen of Indian buffaloBubalusbubalis[J]. Gene, 2012,507(2): 146-151.
[14] Reddy B, Singh K M, Patel A K,etal.. Insights into resistome and stress responses genes inBubalusbubalisrumen through metagenomic analysis[J]. Mol. Biol. Rep., 2014,41(10): 6405-6417.
[15] Pitta D W, Kumar S, Veiccharelli B,etal.. Bacterial diversity associated with feeding dry forage at different dietary concentrations in the rumen contents of Mehshana buffalo (Bubalusbubalis) using 16S pyrotags[J]. Anaerobe, 2014,25: 31-41.
[16] Patel D D, Patel A K, Parmar N R,etal.. Microbial and carbohydrate active enzyme profile of buffalo rumen metagenome and their alteration in response to variation in the diet[J]. Gene, 2014,545(1): 88-94.
[17] Parmar N R, Solanki J V, Patel A B,etal.. Metagenome of mehsani buffalo rumen microbiota: An assessment of variation in feed-dependent phylogenetic and functional classification[J]. J. Mol. Microbiol. Biotechnol., 2014,24(4): 249-261.
[18] Roggenbuck M, Sauer C, Poulsen M,etal.. The giraffe (Giraffacamelopardalis) rumen microbiome[J]. FEMS Microbiol. Ecol., 2014,90(1): 237-246.
[19] Jami E, Israel A, Kotser A,etal.. Exploring the bovine rumen bacterial community from birth to adulthood[J]. ISME J., 2013,7(6): 1069-1079.
[20] Palmer C, Bik E M, DiGiulio D B,etal.. Development of the human infant intestinal microbiota[J]. PLoS Biol., 2007,5(7): e177.
[21] Koenig J E, Spor A, Scalfone N,etal.. Succession of microbial consortia in the developing infant gut microbiome[J]. Proc. Natl. Acad. Sci. USA, 2011,108(S1): 4578-4585.
[22] Ilmberger N, Gullert S, Dannenberg J,etal.. A comparative metagenome survey of the fecal microbiota of a breast-and a plant-fed Asian elephant reveals an unexpectedly high diversity of glycoside hydrolase family enzymes[J]. PLoS ONE, 2014,9(9): e106707.
[23] Ziemer C J. Broad diversity and newly cultured bacterial isolates from enrichment of pig feces on complex polysaccharides[J]. Microbial Ecol., 2013,66(2): 448-461.
[24] Ziemer C J. Newly cultured bacteria with broad diversity isolated from eight-week continuous culture enrichments of cow feces on complex polysaccharides[J]. Appl. Environ. Microbiol., 2014,80(2): 574-585.
[25] de Oliveira M N V, Jewell K A, Freitas F S,etal.. Characterizing the microbiota across the gastrointestinal tract of a Brazilian Nelore steer[J]. Vet. Microbiol., 2013,164(3): 307-314.
[26] Ni J J, Yan Q Y, Yu Y H,etal.. Factors influencing the grass carp gut microbiome and its effect on metabolism[J]. FEMS Microbiol. Ecol., 2014,87(3): 704-714.
[27] Singh K M, Shah T M, Reddy B,etal.. Taxonomic and gene-centric metagenomics of the fecal microbiome of low and high feed conversion ratio (FCR) broilers[J]. J. Appl. Genet., 2014,55(1): 145-154.
[28] López-Cortés N, Reyes-Duarte D, Beloqui A,etal.. Catalytic role of conserved HQGE motif in the CE6 carbohydrate esterase family[J]. FEBS Lett., 2007,581(24): 4657-4662.
[29] Palackal N, Lyon C S, Zaidi S,etal.. A multifunctional hybrid glycosyl hydrolase discovered in an uncultured microbial consortium from ruminant gut[J]. Appl. Microbiol. Biotechnol., 2007,74(1): 113-124.
[30] Weimer P J, Stevenson D M. Isolation, characterization, and quantification ofClostridiumkluyverifrom the bovine rumen[J]. Appl. Microbiol. Biotechnol., 2012,94(2): 461-466.
[31] Liu K, Wang J, Bu D,etal.. Isolation and biochemical characterization of two lipases from a metagenomic library of China Holstein cow rumen[J]. Biochem. Biophys. Res. Commun., 2009,385(4): 605-611.
[32] Wang M, Chen F, Zhang S,etal.. Analysis of xylanases derived from the metagenomic BAC clone library of yak rumen [J]. Acta Microbiol. Sin., 2011,51(10): 1364-1373.
[33] Larsbrink J, Rogers T E, Hemsworth G R,etal.. A discrete genetic locus confers xyloglucan metabolism in select human gutBacteroidetes[J]. Nature, 2014,506(7489): 498-502.
[34] Pope P B, Mackenzie A K, Gregor I,etal.. Metagenomics of the Svalbard reindeer rumen microbiome reveals abundance of polysaccharide utilization loci[J]. PLoS ONE, 2012,7(6): e38571.
[35] Gruninger R J, Gong X, Forster R J,etal.. Biochemical and kinetic characterization of the multifunctional beta-glucosidase/beta-xylosidase/alpha-arabinosidase, Bgxa1[J]. Appl. Microbiol. Biotechnol., 2014,98(7): 3003-3012.
[36] Li Y D, Liu N, Yang H,etal.. Cloning and characterization of a new beta-glucosidase from a metagenomic library of rumen of cattle feeding with miscanthus sinensis[J]. BMC Biotechnol., 2014, doi: 10.1186/1472-6750-14-85.
