TDDFT Study on Excited-State Hydrogen Bonding of 2′-Deoxyguanosine in H2O Solution
2015-01-20DonglinLiHuiLiYonggangYangYufangLiu
Dong-lin Li,Hui Li,Yong-gang Yang,Yu-fang Liu
Department of Physics,Henan Normal University,Xinxiang 453007,China
(Dated:Received on April 28,2015;Accepted on July 14,2015)
TDDFT Study on Excited-State Hydrogen Bonding of 2′-Deoxyguanosine in H2O Solution
Dong-lin Li,Hui Li,Yong-gang Yang,Yu-fang Liu∗
Department of Physics,Henan Normal University,Xinxiang 453007,China
(Dated:Received on April 28,2015;Accepted on July 14,2015)
The mono and dihydrated complexes of 2′-deoxyguanosine have been used to elucidate the importance of the 2′-hydroxy group in the hydration.Density functional theory and timedependent density functional theory methods were performed to investigate the groundand excited-state hydrogen bonding properties of 2′-deoxyguanosine-water(2′-dG-W)and 2′-deoxyguanosine-2water(2′-dG-2W).Infrared spectra,geometric optimizations,frontier molecular orbitals and Mulliken charges have also been studied.The results demonstrated that the excited-state intramolecular hydrogen bonding dynamics of complexes 2′-dG-W and 2′-dG-2W behaves differently upon photoexcitation,while their intermolecular hydrogen bonding dynamics behaves similarly.Moreover,the significant weakening of the intermolecular hydrogen bond O4··H1−N1 and the formation of the new strong hydrogen bond O4···H3−N2 in the 2′-dG-2W upon photoexcitation were due to the geometric structure bending of guanine and the rigidity of related molecules.In addition,the charge transfer properties were theoretically investigated by analysis of molecular orbital.
Hydrate,Time-dependent density functional theory,Rigidity,Hydrogen bonding dynamics
I.INTRODUCTION
As the focal points of solution chemistry,the solutesolvent interactions play a fundamental role for molecular non-equilibrium processes in liquids[1−16].The intermolecular hydrogen bonding is one of the most important interactions governing reactivity and chemical structure in fields such as chemistry and molecular biology.It also has a remarkable influence on the photophysics and photochemistry of chromophores in the hydrogen-bonding surroundings[17−31].The hydrogen bonding effects on the structures and dynamics ofmany important molecular systems have been well investigated[9−16].The hydrogen bonding affected the internal conversion,electronic spectral shifts,photoinduced electron transfer,fluorescence quenching,intramolecular charge transfer and metal-to-ligand charge transfer [31,32].However,the nature of hydrogen bond and its impact factors upon photo-excitation have not been fully explained up to now.
It is well known that the molecular photochemistry in solution is of fundamental importance in modern chemistry and is affected significantly by intermolecular interactions between the solute and the solvent.Intermolecular hydrogen bonds are very important types of solute-solvent interaction and have been recognized to play a fundamental role in molecular and supramolecular photochemistry[33−43].The 2′-deoxyguanosine(2′-dG)is a reverse transcriptase inhibitor and has a strong activity for the prevention of hepatitis virus[44].Therefore,hydrate nature study of 2′-dG has very important significance.But few studies reported the nature of the intermolecular hydrogen bonds and the intramolecular hydrogen bond of hydrates of 2′-dG after photo-excitation,only the most stable conformation of 2′-deoxyguanosine-water (2′-dG-W)and 2′-deoxyguanosine-2water(2′-dG-2W) complexes have been studied[45].
In this work,based on the stable conformation of 2′-dG-W and 2′-dG-2W,a comprehensive investigation of the intramolecular and intermolecular hydrogen bonding of both 2′-dG-W and 2′-dG-2W in excited states were performed,which is necessary to better understand the properties of 2′-dG-Wand 2′-dG-2Wcomplexes.The excited-state geometric optimizations have been performed.The electronic excitation energies,oscillation strengths,and infrared spectra of the electronically excited states were studied to deeply analyze the properties of hydrogen bonding.The Mulliken charges and frontier molecular orbitals were studied to give a more clear view of hydrogen bonds changes in excited states.The influences of the rigidity of the guanine moiety on hydrogen bonds were also investigated.
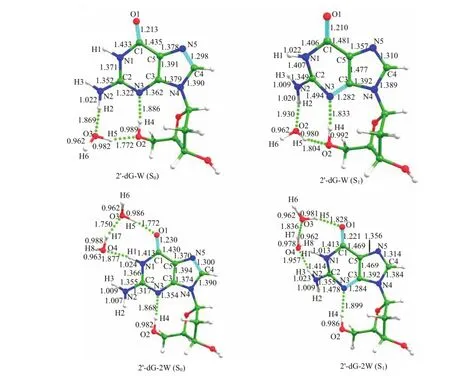
FIG.1 The optimized geometric structures of 2′-dG-2W in states S0and S1.Some important bond lengths are labeled. The geometric structures of 2′-dG-W in states S0and S1is also list for comparison.
II.THEORETICAL METHOD
The geometry optimizations for the ground state and the first singlet electronic excited state were performed using density functionaltheory(DFT)and timedependent density functional theory(TDDFT)methods,respectively.B3LYP functional(Becke’s threeparameter hybrid exchange function with Lee-Yang-Parr gradient-corrected correlation functional)was used in both the DFT and TDDFT methods[46−48].The triple-ζvalence quality with one set ofpolarization functions(TZVP)was chosen as the basis set in all the calculations[49].The TDDFT/B3LYP was used to calculate the excited states with the TZVP basis set. TDDFT was also chosen as it has been confirmed as a reliable method to study the excited state hydrogen bonding dynamics.Fine quadrature grids of size 4 were employed[50].The self-consistent field convergency optimization were set to be 10−8(default settings are 10−6).All the calculated vibrational frequencies were scaled by the factor of 0.961 to correct the anharmonicity effects in this work[51].All the electronic structure and spectra calculations were carried out using the TURBOMOLE program suit[46−51]. The water was used as solvent by using the conductorlike screening model method in order to evaluate the solvent effect[52].
III.RESULTS AND DISCUSSION
Acting well as an important hydrogen donor and acceptor,water plays a key role in weak noncovalent interactions of some vital processes[53].Moreover,the hydration plays a major role in the stabilization of DNA and RNA[54].In this work,the dihydrate of 2′-dGwas discussed in detail,and the monohydrate was also studied for comparison.
Figure 1 shows the optimized geometric structures of monohydrated(2′-dG-W)and dihydrated(2′-dG-2W)complexes.For 2′-dG-W,the six-numbered cycle of the guanine moiety is in the plane,and it bends a lot after being excited to the S1state.The angle∠N2C2C5 changes to 155◦in the S1state from 175◦in the ground state.This change can be explained by the charge distribution of different moieties beforeand after photoexcitation.The intramolecular hydrogen bond N3···H4−O2 in ground state is calculated as 1.886˚A,and is shortened to 1.833˚A in the S1state,which indicates that the intramolecular hydrogen bond is strengthen.For 2′-dG-2W,upon photoexcitation,the intermolecular hydrogen bond interaction sites change from O2−H4 and N2−H2 groups to C1=O1 and N1−H1 groups,respectively.The bond length of intermolecular hydrogen bond C1=O1···H5 increases by 0.056˚A.Meanwhile,the intermolecular hydrogen bonding O4−H7··O3 formed between two waters is lengthened by 0.086˚A in the S1state.More importantly, it is interesting to find that the intermolecular hydrogen bond O4··H1−N1 in the S1state is significantly weaken to 3.021˚A and the new strong hydrogen bond O4···H3−N2 is formed.This should be due to the geometric structure bending of guanine.That is to say, the formation and weakened ofhydrogen bonds between two or more molecules are influenced by the rigidity of related molecules.Moreover,the bond length of new formed hydrogen bonding O4··H3−N2 is calculated to be 2.422˚A in SOstate and shortened to 1.957˚A in the S1state.Therefore,it can be concluded that the intermolecular hydrogen bonds C1=O1··H5 and O4−H7··O3 are weakened after photoexcitation,while the new formed hydrogen bond O4··H3−N2 is strengthened.The intramolecular hydrogen bond N3··H4−O2 in 2′-dG-2Wincreases by 0.031˚A and is weakened after photoexcitation,whose excited state hydrogen bonding dynamics behavior is different from complex 2′-dG-W.
The electronic excitation energies,oscillator strengths,and frontier molecular orbitals transition for the first six excited states of 2′-dG,2′-dG-W, and 2′-dG-2W are provided in Table I.It can be seen that the oscillator strengths of the S1and S2states are much larger than those of the other four excited states.In this work,we focus mainly on the S1state,a fluorescent state.For the monohydrated complex 2′-dG-W and dihydrated 2′-dG-2W,the electronic excitation energies of the S1state are both calculated to be 253 nm.It can be found that the electronic excitation energy of 2′-dG hardly changes compared to its monohydrated complex 2′-dG-W and dihydrated complex 2′-dG-2W.That is to say, the formation of intermolecular hydrogen bonding of 2′-dG-W and 2′-dG-2W has little influence on the electronic absorption spectra.
The Mulliken charge population analysis can provide a means of estimating partial atomic charges from calculations carried out by the methods of computational chemistry.To obtain more information regarding the distribution of partial atomic charges,the charges of 2′-dG-W and 2′-dG-2W are provided in Fig.2.For 2′-dG-W,the charge of N3 atom in ground state is calculated to be−0.285,and changes to−0.283 in the S1state.The charge of H4 atom decreases from 0.335 in ground state to 0.307 in the S1state.That is to say the charge of N3 atom becomes less negative and the chargeof H4 atom becomes less positive.So the intramolecular hydrogen bonding N3··H4−O2 is weakened upon photo-excitation.This is inconsistent with the above geometric structure analysis,which demonstrates that the intramolecular hydrogen bonding N3···H4−O2 is strengthened in S1state.This inconsistency should be due to the geometric structure bending of six-numbered cyclic guanine moiety.For 2′-dG-2W,there are three intermolecular hydrogen bonds and one intramolecular hydrogen bond formed in the ground state.Firstly,the charges of N3 and H4 are calculated to be−0.271 and 0.314 in ground state,which change to−0.236 and 0291 respectively upon photoexcitation.This indicates that the intramolecular hydrogen bond interaction is weakened after excitation.Secondly,the charges of O1 and H5 are calculated to be−0.415 and 0.308 in ground state,which change to−0.358 and 0.304 in the S1state, respectively.That is to say,the atom O1 and atom H5 are less attractive.Therefore,the intermolecular hydrogen bond interaction is weakened.Thirdly,similar to that of H5··O1−C1,the change of O3··H7−O4 is weakened in the S1state.Finally,the charges of atoms O4 and H1 change from−0.594 and 0.265 in ground state to−0.585 and 0.243 in the S1state,respectively. This dramatic change indicates that the hydrogen bond O4··H1−N1 is significantly weakened in the S1state.
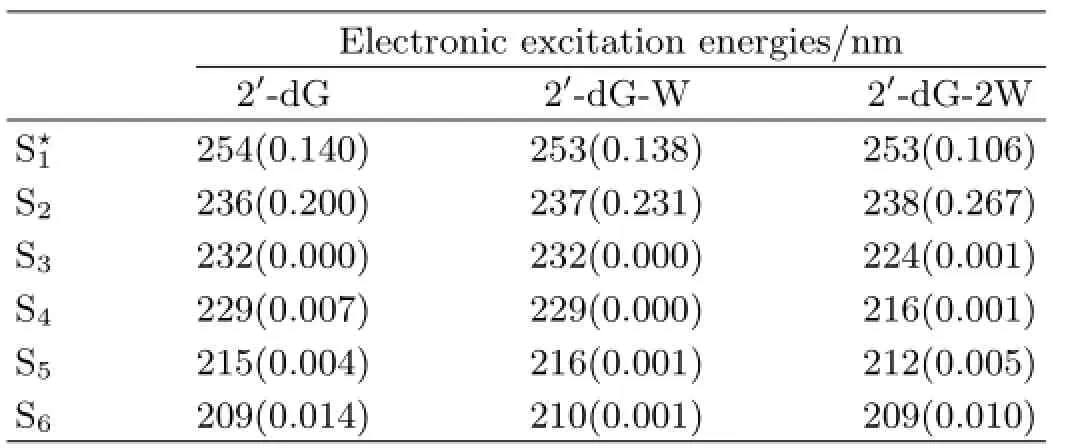
TABLE I The calculated electronic excitation energy and corresponding oscillator strengths(in parentheses)of monomer 2′-dG(keto)as well as the mono-and dihydrates complexes.
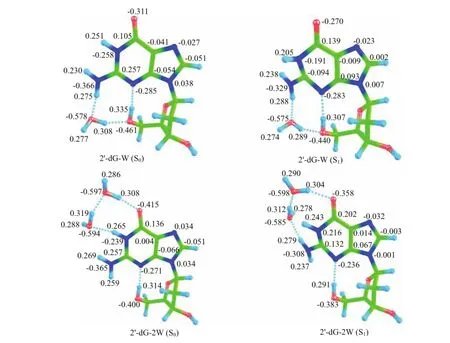
FIG.2 The Mulliken charges of atoms related to hydrogen bonds of 2′-dG-W and 2′-dG-2W in the ground and excited S1states.
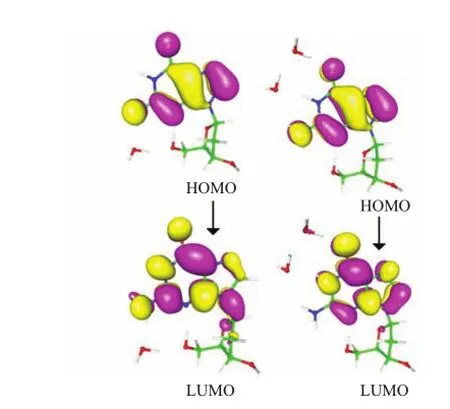
FIG.3 The frontier molecular orbitals of2′-dG-W(left)and 2′-dG-2W(right)in the excited S1state.
The frontier molecular orbitals are carried out to obtain information regarding the electron density of the mono-and dihydrates water complexes.The analysis of molecular orbitals was found to provide the key to the understanding of the electronic structure of atoms and molecular systems[55].According to the data provided in Table I,the S1states of2′-dG-Wand 2′-d G-2W all correspond to the transition from the highest occupied molecular orbital(HOMO)to the lowest unoccupied molecular orbital(LUMO).Therefore,only the frontier molecular orbitals HOMO and LUMO of monoand dihydrates complexes are shown in Fig.3.In Fig.3, the HOMO and LUMO of 2′-dG-Wand 2′-d G-2Whave πandπ∗character,respectively.For 2′-dG-W,however,the electron density of N3 atom has lessπ∗character,which is inconsistent with the strengthening of intramolecular hydrogen bonding N3··H4−O2.This inconsistency should be caused by the influence of the two intermolecular hydrogen bonds and the geometric structure bending of guanine moiety.For 2′-dG-2W, the electron density of N3 atom decreases obviously in LUMO orbital,which consists with the weakening of hydrogen bonding interaction N3··H4−O2.In addition, it is obvious that the electron density of N1−H1 group increases significantly,while that of N2−H3 decreases sharply.This dramatic change indicates the hydrogen bond O4···H1−N1 is significantly weakened and a new hydrogen bond O4··H3−N2 is formed.

FIG.4 The infrared spectra of 2′-dG-W and 2′-dG-2W in the ground S0and the excited S1states.
In order to delineate the change of the intermolecular and intramolecular hydrogen bonding upon photoexcitation,the infrared spectra of 2′-d G-W and 2′-d G-2W are shown in Fig.4.Figure 4(b)also shows the data of corresponding experimental results[45].For 2′-dG-W, the calculated stretching mode of group O2−H4 is at 3185 cm−1in the ground state,while it red-shifts to 3131 cm−1in the S1state.This is in good agreement with the experimental results.The N2−H2 stretching vibration is blue-shifted by 57 cm−1in the S1state.That is to say,the intermolecular hydrogen bond N2−H2··O3 is weakened due to the photoexcitation.For 2′-dG-2W,the H4−O2 stretching vibrational frequency in ground state is 3304 cm−1and is red-shifted by 14 cm−1in the S1state.This indicates the intramolecular hydrogen bond is strengthened when excited to the S1state.The calculated O3−H5 stretching vibrationalfrequency in ground state is 3294 cm−1,while it red-shifts to 3346 cm−1in the S1state.This demonstrates the intermolecular hydrogen bond interaction H5··O1−C1 is weakened upon photoexcitation.By analysis of the H7−O4 stretching vibrational frequency before and after photoexcitation,it is found that the stretch vibration of the intermolecular hydrogen bond interaction O3···H7−O4 is weakened in the S1state.More interestingly,the stretch vibration of N1−H1 is significantly blue-shifted from 3250 cm−1in ground state to 3403 cm−1in the S1state. The large blue-shift 153 cm−1for the N1-H1 stretching mode suggests that the intermolecular hydrogen bond O4··N1−H1 is significantly weakened.In contrast,the stretch vibration of N2−H3 red-shifts 180 cm−1from 3426 cm−1in ground state to 3246 cm−1in S1state. This dramatic shift indicates the intermolecular hydrogen bond O4··H3−N2 is formed in the S1state.The results provide more proofs for the significantly weakened intermolecular hydrogen bond O4··N1−H1 and the formation of O4··H3−N2 in the S1state.
IV.CONCLUSION
In this work,the dihydrate of 2′-dG was discussed and the monohydrate was also studied for comparison. It was demonstrated the guanine moiety of monomer 2′-dG bends considerably when being excited to the S1state.Upon photoexcitation,the intramolecular hydrogen bond N3··H4−O2 of 2′-dG-W is strengthened while that of 2′-dG-2W is weakened.It can be inferred that the change of intramolecular hydrogen bond N3··H4−O2 before and after photoexcitation is not only related to the rigidity of the guanine moiety,but also influenced by the intermolecular hydrogen bonds.Moreover,the intermolecular hydrogen bonds N2−H2··O3 and O3−H5··O2 of 2′-dGW are both weakened after photoexcitation to the S1state.Similarly,the intermolecular hydrogen bonds O3··H7−O4 and H5··O1−C1 of 2′-dG-2W are weakened upon photoexcitation.In addition,it is interesting to find that the intermolecular hydrogen bond O4··H1−N1 of 2′-dG-2Wformed in ground state is significant weakened and a new intermolecular hydrogen bond O4··H3−N2 is formed in the S1state.Therefore,it can be concluded that the charge redistribution after photoexcitation influences the molecules geometric structure as well as the hydrogen bond interaction between solvent and solute molecules.
V.ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China(No.11274096), the Innovation Scientists and Technicians Troop Construction Projects of Henan Province of China (No.124200510013),and the Science and Technology Research Key Project of Education Department of Henan Province of China(No.13A140690).
[1]K.L.Han and G.J.Zhao,Hydrogen Bonding and Transf er in the Excited State,Chichester,UK:John Wiley&Sons Ltd.,(2010).
[2]S.Ghosh,U.Mandal,A.Adhikari,S.Dey,and K.Bhattacharyya,Int.Rev.Phys.Chem.26,421(2007).
[3]E.Pines,D.Pines,Y.Z.Ma,and G.R.Fleming, ChemPhysChem 5,1315(2004).
[4]G.J.Zhao,B.H.Northrop,P.J.Stang,and K.L.Han, J.Phys.Chem.A 114,9007(2010).
[5]M.K.Krepps,S.Parkin,and D.A.Atwood,Cryst. Growth Des.1,291(2001).
[6]C.Graeme and V.M.Rotello,Chem.Soc.Rev.31,275 (2002).
[7]M.A.Walters,C.L.Roche,A.L.Rheingold,and S. W.Kassel,Inorg.Chem.44,3777(2005).
[8]G.A.Jeffrey,An Introduction to Hydorgen Bonding, University Offices,Wellington Square,Oxford OX1 2JD,USA,Oxford University Press,(1997).
[9]G.J.Zhao and K.L.Han,J.Phys.Chem.A 111,2469 (2007).
[10]Y.F.Liu,Y.G.Yang,K.Jiang,D.Shi,and J.Sun, Phys.Chem.Chem.Phys.13,15299(2011).
[11]G.J.Zhao,B.H.Northrop,K.L.Han,and P.J.Stang, J.Phys.Chem.A 114,9007(2010).
[12]G.J.Zhao and K.L.Han,ChemPhysChem 9,1842 (2008).
[13]Y.F.Liu,D.P.Yang,and D.H.Shi,J.Comput.Chem. 32,3475(2011).
[14]A.D.Buckingham,J.E.Del Bene,and S.A.C.Mc-Dowell,Chem.Phys.Lett.463,1(2008).
[15]T.Jorgensen,H.Gardsvoll,and M.Ploug,J.Am. Chem.Soc.127,2785(2005).
[16]G.J.Zhao,K.L.Han,and P.J.Stang,J.Chem.Theory Comput.5,1955(2009).
[17]E.V.Bakhmutova,V.I.Bakhmutov,N.V.Belkova, M.Besora,L.M.Epstein,A.Lled´os,G.I.Nikonov,E. S.Shubina,J.Tom`as,and E.V.Vorontsov,Chem.Eur. J.10,661(2004).
[18]G.J.Zhao and K.L.Han,Phys.Chem.Chem.Phys. 12,8914(2010).
[19]R.Wang,C.Hao,P.Li,N.N.Wei,J.Chen,and J. Qiu,J.Comput.Chem.31,2157(2010).
[20]S.Mukherjee,S.Majumdar,and Bhattacharyya,J. Phys.Chem.B 109,10484(2005).
[21]G.J.Zhao,R.K.Chen,M.T.Sun,J.Y.Liu,G.Y.Li, Y.L.Gao,K.L.Han,X.C.Yang,and L.Sun,Chem. Eur.J.14,6935(2008).
[22]G.Y.Li,G.J.Zhao,Y.H.Liu,K.L Han,and G.Z. He,J.Comput.Chem.31,1759(2010).
[23]Y.F.Liu,J.X.Ding,D.H.Shi,and J.F.Sun,J.Phys. Chem.112,6224(2008).
[24]G.J.Zhao and K.L.Han,J.Phys.Chem.A 111,9218 (2007).
[25]P.S.Murthy,J.Chem.Edu.83,1010(2006).
[26]G.J.Zhao,J.Y.Liu,L.C.Zhou,and K.L.Han,J. Phys.Chem.B 111,8940(2007).
[27]Y.F.Liu,J.X.Ding,R.Q.Liu,D.H.Shi,and J.F. Sun,J.Comput.Chem.30,2723(2009).
[28]C.E.H.Dessent and K.M¨uller-Dethlefs,Chem.Rev. 100,3999(2000).
[29]Y.F.Liu,Y.G.Yang,K.Jiang,D.Shi,and J.Sun, Chem.Soc.Jpn.84,191(2011).
[30]H.Li,Y.F.Liu,and Y.G.Yang,J.Photochem.Photobio.A 291,9(2014).
[31]S.Chai and S.L.Cong,Spectrochim.Acta.A 125,67 (2014).
[32]G.J.Zhao and K.L.Han,Accounts Chem.Res.45, 404(2012).
[33]A.C.Hanglow and P.M.Lydyard,Clin.Exp.Immunol. 59,653(1985).
[34]Z.Shields,P.Zenaida,J.S.Murray,and P.Politzer, Int.J.Quantum Chem.110,2823(2010).
[35]R.Lelchuk,A.Cooke,and J.H.L.Playfair,Cellular. Immunology 72,202(1982).
[36]H.Bril,T.W.van den Akker,and L.M.Hussaarts-Odikjk,R.Benner.Cell.Immunol.90,531(1985).
[37]M.Garc´ıa-Arriaga,G.Hobley,and J.M.Rivera,J.Am. Chem.Soc.130,10492(2008).
[38]H.Asami,K.Yagi,M.Ohba,S.H.Urashima,and H. Saigusa,Chem.Phys.20,84(2013).
[39]H.Saigusa,J.Photochem.Photobio.C 7,197(2006).
[40]E.Nir,I.H¨unig,K.Kleinermanns,and M.S.de Vries, ChemPhysChem 5,131(2004).
[41]J.L.Hougland,R.N.Sengupta,Q.Dai,S.K.Deb,and J.A.Piccirilli,Biochemistry 47,7684(2008).
[42]H.Asami,S.H.Urashima,and H.Saigusa,Phys.Chem. Chem.Phys.11,10466(2009).
[43]A.Abo-Riziq,B.O.Crews,I.Compagnon,J.Oomens, G.Meijer,G.V.Helden,M.Kabel´aˇc,Hobza P,and M. S.de Vries,J.Phys.Chem.A 111,7529(2007).
[44]H.Asami,S.H.Urashima,and H.Saigusa,Phys.Chem. Chem.Phys.11,10466(2009).
[45]R.Ahlrichs,M.B¨Ar,M.H¨Aser,H.Horn,and C. K¨olmel,Chem.Phys.Lett.162,165(1989).
[46]A.D.Becke,J.Chem.Phys.98,5648(1993).
[47]B.G.Johnson,P.M.W.Gill,and J.A.Pople,J.Chem. Phys.98,5612(1993).
[48]A.Sch¨afer,C.Huber,and R.Ahlrichs,J.Chem.Phys. 100,5829(1994).
[49]A.P.Scott and L.Radom,J.Phys.Chem.100,16502 (1996).
[50]A.Klamt,J.Phys.Chem.100 3349(1996).
[51]P.Ball,Chem.Rev.452,291(2008).
[52]E.G.Robertson and J.P.Simons,Phys.Chem.Chem. Phys.3,1(2001).
[53]V.Lemaur,M.Steel,D.Beljonne,J.L.Br´edas,and J. Comil,J.Am.Chem.Soc.127,6077(2005)
∗Author to whom correspondence should be addressed.E-mail: yf-liu@htu.cn,Tel./FAX:+86-373-3329297.
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Visualization of Melting of Antiferromagnetic Insulator Phase in Phase-Separated Manganite Film using Magnetic Force Microscopy
- Oxidation of Anatase TiO2(001)(1×4)Surface
- One-Dimensional Scanning of Electronic Wavefunction in Carbon Nanotubes by Molecular Encapsulation
- Chemical Empiricism 2.0 at Age of Big Data:Large-scale Prediction of Reaction Pathways Based on Bond Dissociation Energies
- First-Principles Study of La Doping Effects on the Electronic Structures and Photocatalytic Properties of Anatase TiO2
- Reconstruction of Smoke Plume Concentration Peaks Based on Modified MAX-DOAS Tomography