舒糖络颗粒质量标准研究
2015-01-17杨玉兰任爱农
杨玉兰, 柴 彦, 任爱农, 赵 妍, 张 梅
(1.江苏大学,江苏镇江212013;2.江苏中豪医药有限公司,江苏南京210009;3.江苏省中医药研究院,江苏南京210028)
舒糖络颗粒质量标准研究
杨玉兰1, 柴 彦2, 任爱农3*, 赵 妍3, 张 梅3
(1.江苏大学,江苏镇江212013;2.江苏中豪医药有限公司,江苏南京210009;3.江苏省中医药研究院,江苏南京210028)
目的建立舒糖络颗粒 (葛根、黄连、鬼箭羽、生地、凌霄花)的质量标准。方法采用薄层色谱法对该复方制剂中葛根、黄连、生地、凌霄花进行定性鉴别,并采用高效液相色谱法对主要成分葛根素、盐酸小檗碱进行定量测定。结果薄层色谱斑点清晰,在与对照品或对照药材相应的位置上显相同颜色的斑点,阴性对照无干扰;葛根素和盐酸小檗碱分别在0.053~0.210mg/mL(r=0.999 7)、0.011~0.192mg/mL(r=0.999 5)范围内线性关系良好;加样回收率 (n=6)均值分别为99.2%、101.6%,RSD分别为2.6%、1.1%。结论所建立的方法操作简便,准确度高,重复性好,能全面、有效地控制该制剂的质量。
舒糖络颗粒;葛根素;盐酸小檗碱;薄层鉴别;高效液相;质量标准
舒糖络颗粒是由江苏省中医药研究院自主研发的科研制剂,源于该院内分泌科临床经验方,由葛根、黄连、鬼箭羽、生地、凌霄花五味中药组成,具有活血通络、凉血祛瘀、滋阴清热的功效,临床上用于治疗糖尿病周围神经病变[1]。糖尿病周围神经病变是糖尿病三大慢性并发症之一。中医理论认为糖尿病周围神经病变属 “络病”范畴,其发病机理为: “脉络瘀滞,不通则痛”和 “脉络空虚,不荣则痛”[2]。现代医学对该病提出了血管障碍和代谢紊乱学说。血管障碍理论认为,长期高血糖可导致血液流变异常,神经微血管内皮增生,管腔变窄,同时血液黏滞度增高,神经内滋养血管易发生堵塞而造成神经营养障碍和变性;代谢障碍学说包括山梨醇、肌醇代谢异常和蛋白非酶糖基化等理论或假说[3]。近代药理研究表明该方各药分别具有改善血液循环和微血管病变,纠正代谢障碍的作用[4-8],为了便于服用和携带,我们将该方采用现代工艺制成稳定的中药制剂,同时为了更加全面控制该制剂的内在质量,保证临床用药的安全有效,本实验采用薄层色谱法对制剂中葛根、黄连、生地、凌霄花四味药材进行了较全面的定性鉴别,并采用高效液相色谱法对葛根素及黄连中的生物碱成分进行了定量测定。
1 仪器与试药
高效液相色谱仪(美国Waters公司,2489紫外/可见检测器,Empower色谱工作站);AT201梅特勒十万分之一电子分析天平(瑞士METTLER公司);KQ-250E医用超声波清洗器 (昆山超声仪器有限公司);硅胶G板 (青岛海洋化工厂分厂);薄层紫外检测仪(瑞士CAMAG公司);Milli-Q超纯水系统(美国Millipore公司)。
葛根素对照品 (批号110752-200912,纯度96.0%)、盐酸小檗碱对照品 (批号713-9204,纯度98.14%)、梓醇对照品 (批号110808-200305,纯度98.1%)、凌霄花对照药材 (批号121122-200402)均购自中国食品药品检定研究院,舒糖络颗粒 (江苏省中医药研究院自制,批号分别为20140102、20140307、20140522)。
黄连、葛根、生地、凌霄花药材购自安徽丰原铜陵中药饮片有限公司,经江苏省中医药研究院任爱农研究员鉴定为毛茛科植物黄连Coptis chinensis Franch.的干燥根茎、豆科植物野葛Pueraria lobata(Willd.)Ohwi的干燥根、玄参禾斗植物地黄Rehmannia glutinosa Libosch.的干燥块根和紫葳科植物凌霄Campsis grandiflora(Thunb.)K.Schum.的干燥花。
甲醇、乙腈为色谱纯,水为超纯水,其余试剂均为分析纯。
2 薄层色谱鉴别
2.1 葛根薄层鉴别 取本品2 g,研细,加甲醇10 mL,超声处理30 min,过滤,滤液水浴蒸干,加甲醇2 mL溶解残渣,作为供试品溶液;取葛根药材粉末1 g,同法制成药材对照品溶液;再取处方中除葛根外的其他原比例各药材,按制备工艺方法制得阴性颗粒样品,取阴性颗粒样品同法制成阴性对照溶液;另称取葛根素对照品,加甲醇制成0.15 mg/mL的溶液,作为对照品溶液。照 《中国药典》2010年版附录ⅥB薄层色谱法项下方法试验,分别吸取上述溶液各5μL,点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水 (6:4:0.7)为展开剂,预饱和30 min后展开,取出,晾干,在紫外光 (λ=365 nm)下观察。3批供试品色谱在与药材对照品色谱、葛根素对照品色谱相应位置上均显示一组相同的蓝色荧光斑点,阴性对照色谱中未见此斑点。见图1。

图1 葛根薄层色谱图Fig.1 TLC chromatogram of Puerariae Radix
2.2 黄连薄层鉴别 取本品2 g,研细,加甲醇25 mL,超声处理30 min,过滤,滤液水浴蒸干,加甲醇2 mL溶解残渣,作为供试品溶液;取黄连药材粉末1 g,同法制成药材对照品溶液;再取处方中除黄连外的其他原比例各药材,按制备工艺方法制得阴性颗粒样品,取阴性颗粒样品同法制成阴性对照溶液;另称取盐酸小檗碱对照品,加甲醇制成0.10 mg/mL的溶液,作为对照品溶液。照《中国药典》2010年版附录ⅥB薄层色谱法项下方法试验,分别吸取上述溶液各5μL,点于同一硅胶G薄层板上,以正丁醇-冰醋酸-水 (7:1:2)为展开剂,预饱和30 min后展开,取出,晾干,在紫外光 (λ=365 nm)下观察。3批供试品色谱在与药材对照品色谱、盐酸小檗碱对照品色谱相应位置上均显示一组相同的黄色荧光斑点,阴性对照色谱中未见此斑点。见图2。
2.3 生地薄层鉴别 取本品2 g,研细,加甲醇20 mL回流提取1 h,放冷,过滤,滤液水浴蒸干,加水5 mL溶解残渣,再用水饱和正丁醇振摇提取5次,每次10 mL,合并正丁醇液,减压回收正丁醇液至无醇味,浓缩液水浴蒸干,加甲醇10mL溶解残渣,作为供试品溶液;取生地药材粉末2 g,同法制成药材对照品溶液;再取处方中除生地外的其他原比例各药材,按制备工艺方法制得阴性颗粒样品,取阴性颗粒样品同法制成阴性对照溶液;另称取梓醇对照品,加甲醇制成0.50 mg/mL的溶液,作为对照品溶液。照 《中国药典》2010年版附录ⅥB薄层色谱法项下方法试验,分别吸取上述溶液各5μL,点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水 (6:4:1)为展开剂,预饱和30 min后展开,取出,晾干,喷以新鲜配制的1%的茴香醛试液,于105℃下加热3~5 min。3批供试品色谱在与药材对照品色谱、梓醇对照品色谱相应位置上显示一组相同的浅紫色斑点,阴性对照色谱中未见此斑点。见图3。
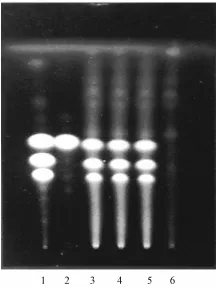
图2 黄连薄层色谱图Fig.2 TLC chromatogram of Coptidis Rhizome
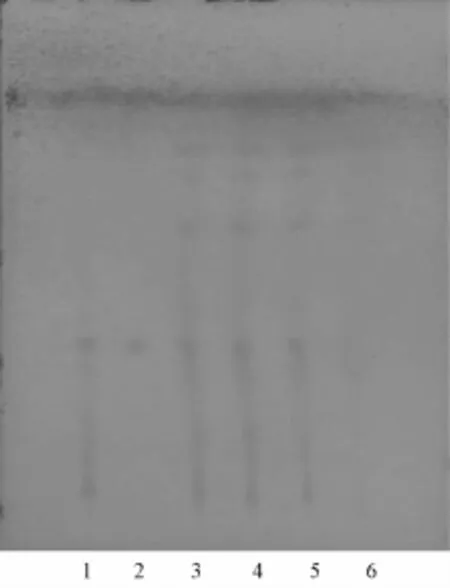
图3 生地薄层色谱图Fig.3 TLC chromatogram of Rehmanniae glutinosa
2.4 凌霄花薄层鉴别 取本品0.3 g,研细,加石油醚 (60~90℃)15 m L,超声处理15 min,过滤,弃去石油醚液,药渣蒸干,加甲醇15 mL,超声处理15 min,过滤,滤液水浴蒸干,加甲醇1 mL溶解残渣,作为供试品溶液;取凌霄花药材粉末0.5 g,同法制成药材对照品溶液;再取处方中除凌霄花外的其他原比例各药材,按制备工艺方法制得阴性颗粒样品,取阴性颗粒样品同法制成阴性对照溶液;另取凌霄花对照药材,同法制成对照药材溶液;照 《中国药典》2010年版附录Ⅵ B薄层色谱法项下方法试验,分别吸取上述溶液各8μL,点于同一硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲醇 (4:4:1)为展开剂,预饱和30 min后展开,取出,晾干,置碘蒸气中熏至斑点显色清晰。3批供试品色谱在与药材对照品色谱、对照药材色谱相应位置上显示一组相同的棕色斑点,阴性对照色谱中未见该组斑点。见图4。
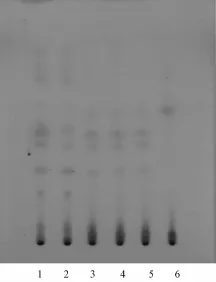
图4 凌霄花薄层色谱图Fig.4 TLC chromatogram of Campsis flos
3 HPLC法测定样品中成分
3.1 葛根素测定
3.1.1 色谱条件 Thermo C18色谱柱(250 mm× 4.6 mm,5μm);流动相为甲醇-水 (22:78),体积流量1.0 mL/min;柱温30℃;检测波长λ=250 nm;进样量10μL。按葛根素峰计算理论塔板数为4 250。
3.1.2 溶液制备
3.1.2.1 对照品溶液制备 精密称取葛根素对照品1.09 mg,以30%乙醇作为溶剂,定容至10 m L,配制成0.105 mg/mL的葛根素对照品溶液。
3.1.2.2 供试品溶液制备 取批号20140522的舒糖络颗粒适量,粉碎。取细粉0.2 g,精密称定,加30%乙醇20 m L,称定质量,超声处理 (功率250 W,频率40 kHz)30 min、取出、放至室温后用30%乙醇补足减失质量,摇匀,过滤,取续滤液作为供试品溶液。
3.1.2.3 阴性对照溶液制备 取 “2.1”项下阴性颗粒样品,按 “3.1.2.2”项下制备方法制成葛根阴性对照溶液。
3.1.3 专属性试验 分别取对照品溶液、供试品溶液和阴性对照溶液各10μL,按 “3.1.1”项下色谱条件分别进样,记录色谱图,供试品溶液在与葛根素对照品溶液相同的保留时间处显示相同特征峰,阴性对照溶液无此特征峰,表明该方法专属性良好。见图5。

图5 葛根HPLC图谱Fig.5 HPLC chromatograms of Puerariae Radix
3.1.4 线性关系考察 精密称取葛根素对照品,加入30%乙醇溶液,配制成系列不同质量浓度对照品溶液 (0.210、0.187、0.168、0.140、0.105、0.070、0.053 mg/mL),按 “3.1.1”项下色谱条件进行分析,以峰面积积分值为纵坐标 (Y),进样质量浓度为横坐标 (X),计算得线性回归方程Y=3.76×107X-7.36×104(r=0.999 7),结果表明葛根素在0.053~0.210 mg/mL范围内具有良好的线性关系。
3.1.5 检测限 取葛根素对照品溶液按倍比稀释法制备测定溶液,按 “3.1.1”项下色谱条件测定,以信噪比S/N=3:1计,葛根素最低检测质量浓度为0.214μg/mL。
3.1.6 定量限 取葛根素对照品溶液按倍比稀释法制备测定溶液,按 “3.1.1”项下色谱条件测定,以信噪比S/N=10:1计;平行6份。葛根素最低检测质量浓度平均为0.535μg/mL,其RSD为1.2%。
3.1.7 精密度试验 吸取对照品溶液10μL,按 “3.1.1”项下色谱条件连续进样6次,测定葛根素对应吸收峰面积。其平均峰面积积分值为3 871 803,其RSD为0.3%,表明仪器精密度良好。
3.1.8 重复性试验 取“3.1.2.2”项下舒糖络颗粒细粉0.2 g,按该项下方法制备供试品溶液,平行处理6份,按 “3.1.1”项下色谱条件分别进样,测定葛根素对应吸收峰面积,计算得葛根素平均含有量为9.55 mg/g,其RSD为1.1%,表明该方法重复性良好。
3.1.9 中间精密度 取“3.1.2.2”项下舒糖络颗粒细粉0.2 g,按该项下方法制备供试品溶液;分别在不同人员、实验仪器和色谱柱,按“3.1.1”项下色谱条件测定,计算供试品中葛根素的量,经t检验P值为0.19,P>0.05,表明组间无显著性差异。结果见表1。

表1 葛根素中间精密度试验结果Tab.1 Results of intermediate p recision for puerarin
3.1.10 稳定性试验 取“3.1.2.2”项下舒糖络颗粒细粉0.2 g,按该项下方法制备供试品溶液,在室温 (25℃)条件下,按 “3.1.1”项下色谱条件于制备后0、1、2、4、6、8、12 h分别进样,测定葛根素对应吸收峰面积,计算得葛根素平均含有量为9.41 mg/g,其RSD为0.9%,表明在室温条件下供试品溶液12 h内稳定。
3.1.11 回收率试验 取“3.1.2.2”项下舒糖络颗粒细粉0.1 g,共6份,每份分别精密加入质量浓度为0.200 mg/mL的葛根素对照品溶液5 mL,按 “3.1.1”项下色谱条件分别测定,计算回收率(外标法)。结果见表2。
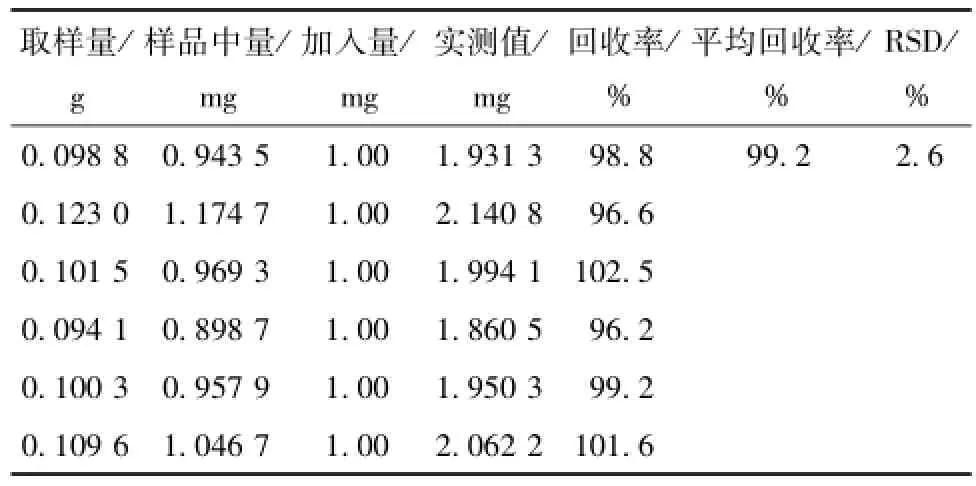
表2 葛根素回收率试验结果Tab.2 Results of recovery tests for puerarin
3.1.12 样品测定 分别取3批舒糖络颗粒样品,按 “3.1.2.2”项下供试品制备方法制备供试品溶液,按 “3.1.1”项下色谱条件分别对3批样品进行测定,计算葛根素量。见表3。

表3 舒糖络颗粒中葛根素测定结果 (n=3)Tab.3 Determ ination results of puerarin in Shutangluo Granules(n=3)
3.2 盐酸小檗碱测定
3.2.1 色谱条件 Thermo C18色谱柱(250 mm× 4.6 mm,5μm);流动相为乙腈-0.05 mol/L磷酸二氢钾 (50:50) (每100 m L流动相中加十二烷基硫酸钠0.4 g,再以磷酸调节pH=4),体积流量1 mL/min;柱温30℃;检测波长λ=345 nm;进样量10μL。按盐酸小檗碱峰计算理论塔板数为11 660。
3.2.2 溶液制备
3.2.2.1 对照品溶液制备 精密称取盐酸小檗碱对照品0.65 mg,以甲醇作为溶剂,定容至10 m L,配制成0.064 mg/mL的盐酸小檗碱对照品溶液。
3.2.2.2 供试品溶液制备 取批号20140522的舒糖络颗粒适量,粉碎。取细粉2.5 g,精密称定,加甲醇-盐酸 (100:1)25 mL,称定质量,超声处理(功率250 W,频率40 kHz)30 min,取出放至室温后用甲醇补足减失质量,摇匀,过滤,取续滤液作为供试品溶液。
3.2.2.3 阴性对照溶液制备 取 “2.2”项下阴性颗粒样品,按 “3.2.2.2”项下制备方法制备黄连阴性对照溶液。
3.2.3 专属性试验 分别取对照品溶液、供试品溶液和阴性对照溶液各10μL,按 “3.2.1”项下色谱条件分别进样,记录色谱图。供试品溶液在与盐酸小檗碱对照品溶液相同的保留时间处,显示相同特征峰,阴性对照溶液无此特征峰,表明该方法专属性良好。见图6。
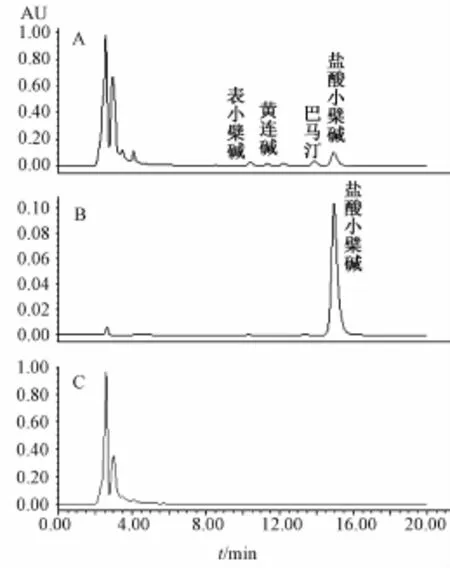
图6 黄连HPLC图Fig.6 HPLC chromatogram s of Coptidis Rhizome
3.2.4 线性关系考察 精密量取盐酸小檗碱对照品,加入甲醇溶液,配制系列不同质量浓度对照品溶 液 (0.192、0.144、0.096、0.064、0.038、0.021、0.011 mg/mL),按 “3.2.1”项下色谱条件进行分析,以峰面积积分值为纵坐标 (Y),进样质量浓度为横坐标 (X),计算得到线性回归方程Y=3.65×107X+5.58×104(r=0.999 5),结果表明盐酸小檗碱在0.011~0.192 mg/mL范围内具有良好的线性关系。
3.2.5 检测限 取葛根素对照品溶液按倍比稀释法制备测定溶液,按 “3.2.1”项下色谱条件测定,以信噪比S/N=3:1计,盐酸小檗碱最低检测质量浓度为0.064μg/mL。
3.2.6 定量限 取葛根素对照品溶液按倍比稀释法制备测定溶液,按 “3.2.1”项下色谱条件测定,以信噪比S/N=10:1计,盐酸小檗碱最低检测质量浓度为0.026μg/mL,其RSD值为2.8%。
3.2.7 精密度试验 吸取对照品溶液10μL,按“3.2.1”项下色谱条件连续进样6次,测定盐酸小檗碱对应吸收峰面积。其平均峰面积积分值为240 850,其 RSD为 0.5%,表明仪器精密度良好。
3.2.8 重复性试验 取“3.2.2.2”项下舒糖络颗粒细粉2.5 g,按该项下方法制备供试品溶液,平行处理6份,按 “3.2.1”项下色谱条件分别进样。测定盐酸小檗碱对应吸收峰面积,计算得盐酸小檗碱平均含有量为0.47mg/g,其RSD为1.6%,表明该方法重复性良好。
3.2.9 中间精密度 取“3.2.2.2”项下舒糖络颗粒细粉2.5 g,按该项下方法制备供试品溶液;分别在不同实验人员、实验仪器和色谱柱的条件下,按 “3.2.1”项下色谱条件测定,计算供试品中盐酸小檗碱的量,其平均含有量分别为0.49和0.47 mg/g,P=0.06,P>0.05,表明组间无显著性差异。见表4。

表4 盐酸小檗碱中间精密度实验结果Tab.4 Results of intermed iate precision for berberine hydrochloride
3.2.10 稳定性试验 取“3.2.2.2”项下舒糖络颗粒细粉2.5 g,按该项下方法制备供试品溶液,在室温 (25℃)条件下,按 “3.2.1”项下色谱条件于制备后0、1、2、4、6、8、12 h分别进样,测定盐酸小檗碱吸收峰面积,计算得盐酸小檗碱平均含有量为0.47 mg/g,其RSD为2.1%,表明室温条件下供试品溶液12 h内稳定。
3.2.11 回收率试验 取“3.2.2.2”项下已知盐酸小檗碱含有量的舒糖络颗粒细粉约1.25 g,共6份,每份中分别精密加入质量浓度为0.122 mg/m L的盐酸小檗碱对照品溶液5 m L,并按照“3.2.2.2”项下方法制备供试品溶液,按“3.2.1”项下色谱条件分别测定,计算回收率(外标法)。见表5。
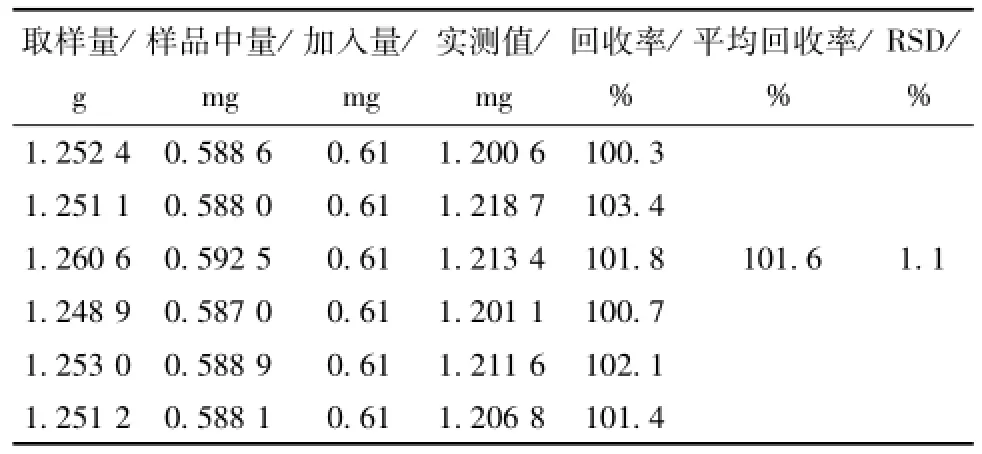
表5 盐酸小檗碱回收率试验结果Tab.5 Results of recovery tests for berberine hydrochloride
3.2.12 样品测定 分别称取3批舒糖络颗粒样品(n=3),按照 “3.2.2.2”项下供试品制备方法制备供试品溶液,按 “3.2.1”项下色谱条件分别对3批样品进行测定,计算盐酸小檗碱量。见表6。
3.2.13 样品中黄连系列生物碱测定 以未知成分的色谱峰与盐酸小檗碱色谱峰的相对保留时间、黄连药材系列生物碱色谱峰的保留时间以及对应光谱图作对照,确定表小檗碱、黄连碱和巴马汀色谱峰,并以盐酸小檗碱对照品的峰面积作对照,分别计算上述3种生物碱的量。见表7,图7。

表6 舒糖络颗粒中盐酸小檗碱测定结果 (n=3)Tab.6 Results of determ ination of Shutangluo Granules(n=3)

表7 样品中其他生物碱含有量Tab.7 Other alkaloid contents from samples

图7 表小檗碱、黄连碱及巴马汀光谱图Fig.7 Spectral patterns of epiberberine,coptisine and palmatine
4 讨论
《中国药典》2010年版规定,梓醇和毛蕊花糖苷同为生地药材的薄层鉴别的指标性成分。但对舒糖络颗粒中梓醇进行薄层鉴别时,前处理仅采用《中国药典》2010年版甲醇回流提取的方法,供试品色谱中色带明显,梓醇斑点模糊,为了对梓醇进行富集和减少其他成分的干扰,本实验考察了用乙酸乙酯、正丁醇和三氯甲烷萃取的方法,结果表明,正丁醇萃取后供试品及药材中梓醇斑点较清晰,故将梓醇鉴别列入正文。采用 《中国药典》2010年版方法对舒糖络颗粒中毛蕊花糖苷进行薄层鉴别时,供试品色谱中未见毛蕊花糖苷的斑点,可能是由于水提取过程中毛蕊花糖苷发生了水解反应,生成5-羟甲基糠醛[9],且为生地和凌霄花的共有成分,故未将毛蕊花糖苷列入鉴别正文。
在对葛根素进行定量测定的过程中,以葛根素提取率为指标,分别对提取方法 (超声、回流)、提取溶剂 (30%、70%和95%乙醇)和提取时间(15、30和45 min)3个因素进行了考察。结果显示,采用30%乙醇提取30 min提取效率较高,而提取方式对提取效率无影响,考虑到超声提取方法更简便,故采用超声提取[10]。在对黄连生物碱进行定量测定中,以盐酸小檗碱提取率为考察指标,分别对提取溶剂 (甲醇-盐酸50:1、100:1、150:1)和超声时间 (15、30和45 min)2个因素进行了考察。结果显示,以甲醇-盐酸 (100:1)为溶剂、超声30 min或45 min盐酸小檗碱提取效率最高,提示超声30 min便可将盐酸小檗碱提取完全。
实验过程中,曾尝试对制剂中已知成分梓醇和毛蕊花糖苷进行定量测定。但是,在测定过程中发现梓醇不稳定,可能是因为处方药材经合煎后pH发生了改变,梓醇中存在的糖苷配体在弱酸条件下易发生水解反应[11]。而毛蕊花糖苷在制剂中含有量极少,同样也很不稳定[9],故未将上述两种成分列入标准。
样品中黄连系列生物碱成分的定位首先参考《中国药典》2010年版一部黄连项下,以待测成分色谱峰与盐酸小檗碱色谱峰的相对保留时间定位,为排除样品中其他药材成分的干扰,同时以黄连药材中的系列生物碱色谱峰的保留时间以及对应光谱图作对照,最终确定了表小檗碱、黄连碱和巴马汀3个生物碱成分色谱峰的位置。
[1]王小超,刘克冕,狄红杰,等.舒糖络方治疗糖尿病周围神经病变的临床观察[J].河北中医,2010,329(1):16-17.
[2]张 兰,姜维娜,陈盛业,等.糖尿病周围神经病变 “络损”病机研究 [C]//第五届国际中医糖尿病大会暨国家中医药糖尿病临床研究联盟成立大会论文集.中华中医药学会,2011:7.
[3]沈稚舟.糖尿病慢性并发症[M].上海:上海医科大学出版社,1999:206-287.
[4]李 莉.生地黄治疗糖尿病的药理研究[J].长春中医药大学学报,2011,27(4):670-672.
[5]李路丹,谢梦洲,赵蒙蒙,等.鬼箭羽对2型糖尿病血瘀证大鼠血糖及血液流变学的影响[J].中南大学学报:医学版,2011,36(2):128-132.
[6]高天曙,梁瑞峰.葛根素对糖尿病大鼠胰岛素、血糖及血液流变学的影响[J].中药药理与临床,2011,27(6):31-33.
[7]张春静.黄连药理作用研究进展概述[J].科技创新与应用,2013(5):101.
[8]吴 伟,朱章志,李 红,等.葛根素治疗早期2型糖尿病肾病的 Meta分析[J].中成药,2013,35(7):1399-1406.
[9]Wang Jing,Kong Hongwei,Yuan Zimin,et al.A novel strategy to evaluate the quality of traditional Chinesemedicine based on the correlation analysis of chemical finge print and biological effect[J].JPharm Biomed Anal,2013(83):57-64.
[10]宋亚芳,苏春梅,杨 红,等.HPLC同时测定葛根芩连微丸中葛根素、甘草苷、黄芩苷、盐酸小檗碱的含量[J].中国实验方剂学杂志,2013,19(2):64-64.
[11]祖勃孙,周 琴.兰考泡桐木材成分的变色行为及其变色过程[J].林业科学,1998,34(3):97.
Quality control of Shutangluo Granules
YANG Yu-lan1, CHAIYan2, REN Ai-nong3*, ZHAO Yan3, ZHANG Mei3
(1.Jiangsu University,Zhenjiang 212013,China;2.Jiangsu Zhonghao Medicine Company,Nanjing 21009,China;3.Jiangsu Provincial Institute of Traditional Chinese Medicine,Nanjing 210028,China)
AIMTo establish the quality control of Shutangluo Granules(Puerariae lobatae Radix,Coptidis Rhizoma,Euonymusalarus,Rehmanniaeglutinosa,CampsisFlos).METHODSTLCwasused to identify Puerariae lobataeRadix,Coptidis Rhizoma,Rehmanniae glutinosa,and Campsis Flos.The quantitative determination ofpuerarin and berberine hydrochloride was completed by HPLC.RESULTSThe spots in TLC were clear without interference.And the same color spots appeared in the corresponding position of references and reference drugs. The linear ranges of puerarin and berberine hydrochloridewere 0.053-0.210 mg/mL(r=0.999 7),and 0.011 -0.192 mg/mL(r=0.999 5),respectively.The average sample recoveries of puerarin,berberine hydrochloride were 99.2%(RSD=2.6%),and 101.6%(RSD=1.1%),respectively.CONCLUSIONThe quality control established is simple,accurate,reproducible and specific,which can be used for the quality control of Shutangluo Granules.
Shutangluo Granules;puerarin;berberine hydrochloride;TLC;HPLC;quality standard
R927.2
A
1001-1528(2015)04-0785-08
10.3969/j.issn.1001-1528.2015.04.020
2014-08-01
江苏省社会发展科技计划、医药高技术计划项目 (BS2007091)
杨玉兰 (1989—),女,硕士生,从事药物新剂型与新技术研究。
*通信作者:任爱农(1957—),女,研究员。Tel:(025)85639647,E-mail:lyyy-0@126.com
