新药萘夫西林药理性质的密度泛函研究
2015-01-10罗海霞钟爱国
徐 洋,罗海霞,范 强, 钟爱国
新药萘夫西林药理性质的密度泛函研究
徐 洋,罗海霞,范 强, 钟爱国
(台州学院医药化工学院, 浙江 台州 316000)
使用密度泛函理论(DFT),在B3LYP方法和6-31+G(d)基组上,对抗菌消炎新药萘夫西林分子进行结构优化,对其分子构型、偶极距、红外光谱、紫外光谱、NBO电荷分布进行了分析。结果表明,萘夫西林发挥其药理和毒理作用的最大可能部位在酰胺基的N原子上。
萘夫西林;密度泛函理论;定量计算
萘夫西林英文名为Nafcilline、Unipen 、Naftopen等,为以 6-氨基青霉素烷酸(6-APA)化学半合成的一种既口服也可注射的新型青霉素[1-3]。为微黄白色或白色粉末,有引湿性,易溶于水、乙醇、氯仿,微臭。在酸和青霉素酶中稳定。主要试用于治疗心内膜炎、急性化脓性的脑膜炎、肺炎、败血症(金黄色葡萄球菌引起)等。与其他青霉素类药物一样的是,它也存在过敏反应。抗耐药金葡菌的活性与苯唑青霉素比较接近,比甲氧苯青霉素要强,对溶血性链球菌和肺炎链球菌的活性比苯唑西林强。特别的是,对溶血性链球菌和肺炎球菌的效果非常好。口服的吸收效果不是很好,尿液排泄量少,血药浓度也较低。通过肌肉注射萘夫西林的血药浓度高于口服,但是仍然比较低,胆汁中的药的浓度高于血药浓度,且90 % 都经胆汁排泄排出。血浆蛋白结合率高,约为88 %。阿拉宾度公司(Aurobindo)在2012年12月29日宣布了萘夫西林注射液(nafcillin injections)已获得了FDA的批准, 该公司也已马上启动了该药物的装运工作。目前,用密度泛函研理论究萘夫西林方面的文献还比较缺乏[4,5]。
本文是采用密度泛函理论方法, 对萘夫西林的分子光谱如红外吸收,拉曼吸收,紫外-可见吸收,核磁共振吸收以及自然键轨道理论(NBO)等进行了理论模拟和指认,通过先采用GaussView 3.7构造出萘夫西林的分子结构,再提交给Gaussian 09W 用相应的方法和基组计算光谱数据,取得了与实验相吻合的结果, 为萘夫西林的分析与检测提供帮助。并结合所得数据,讨论分析了萘夫西林的药理及毒理性质。
1 实验部分
采用GaussView看图软件,构建了萘夫西林分子结构模型,采用 DFT 理论的 B3LYP 方法,在6-31+G(d)基组水平上,进行了结构优化计算。在优化结构的基础上,采用频率分析方法,进行了频率分析。表明所有振动频率全部为正,表明其计算结果是可以相信的。全部计算工作在 Gaussian 09W程序及 PC 机上完成。图 1显示了萘夫西林(C21H22N2O5S)分子结构和原子编号。
2 结果与讨论
2.1 分子的几何结构分析
用Gaussian 09W程序在B3LYP/6-31+G(d)基组上进行优化,模拟得到了萘夫西林分子的键长, 键角等数据。正常的C-O单键的键长((1.20~1.43)×10-10m),C-C单键(1.54×10-10m左右),C=O双键(1.20×10-10m左右),C-N单键(1.48×10-10m),C-S单键(1.82×10-10m),O-H(0.98×10-10m),N-H(1.01×10-10m)表中列出了优化后的分子部分键长数据表明:除C4-C1的键长偏小一点外,其他键长均属于正常范围。表中列出的重要键角数据表明,稠杂环的大环(N1-C2-S6-C7-C3原子组成的五元杂环)和小环(N1-C2-C5-C4原子组成的四元杂环)。

图1 萘夫西林分子结构及编号Fig.1 Structure and atomic of nafcilline
2.2 红外吸收光谱
物质的IR光谱是其分子骨架结构的反映,图2谱图中的吸收峰与分子中各基团振动形式相对应。
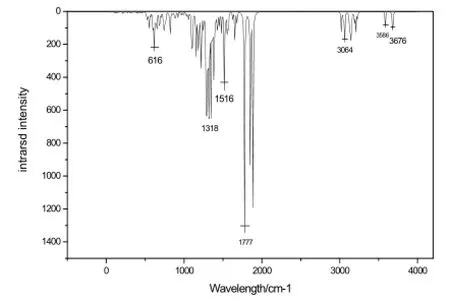
图2 萘夫西林分子红外吸收光谱Fig.2 IR of nafcilline
使用 DFT/B3LYP/6-31+G(d) 软件模拟显示, 萘夫西林药物分子在指纹区 616 cm-1(弱,2S, 5R, 6R结构)处存在红外吸收峰,在特征区1 318 cm-1(中,甲基C-H的弯曲振动吸收峰),1 516 cm-1(弱,萘环上的C-C骨架振动吸收峰), 1 777 cm-1(强,羰基C=O键伸缩振动吸收峰), 3 064 cm-1(弱,萘环上的C-H伸缩振动吸收峰), 3 586 cm-1(弱,萘酰胺基上的N-H伸缩振动吸收峰), 3 676 cm-1(弱,羧基上的羟基O-H的伸缩振动吸收峰)处显示较强的IR吸收峰,这些峰与萘夫西林小分子的实验IR谱(分别在608,1 305,1 556,1 787,3 101,3 576,3 656 cm-1处有红外吸收峰)基本是吻合的。
2.3 拉曼吸收光谱
分子的拉曼吸收效应起源于分子振动与转动,从拉曼吸收光谱中可得到与分子振动互补的信息。采用DFT/B3LYP 方法,在 6-31+G(d)基组上进行分子优化和频率分析,得到7个拉曼吸收特征峰(如图3)。我们对分子的7个特征峰的分子振-转吸收模式进行了归属指认。萘夫西林((2S,5R,6R)-6-(2-乙氧基-1-萘酰氨基)-3,3-二甲基-7-羰基-4-硫杂-1-氮杂-二环[3.2.0]庚烷-2-羧酸)是一个稠杂环的化合物,也可以看作是萘环的β位上氢原子被一个乙氧基所取代,α位上的氢原子被一个由酰胺基和稠杂环构成的支链(青霉素母核-6-氨基青霉烷酸(6-APA))所取代而形成的双取代萘类分子。图 3是理论模拟萘夫西林分子的拉曼散射谱特征吸收峰,其主要位于3 676 cm-1(弱,羧基上的游离羟基O-H的伸缩振动),3 586 cm-1(弱,萘酰胺基上的N-H伸缩振动),3 064 cm-1(强,二取代萘环上的C-H对称呼吸振动),1 777 cm-1(弱,羰基C=O键伸缩振动), 1 516 cm-1(中,萘环上的C-C骨架振动),1 318 cm-1(中,甲基C-H的弯曲振动),616 cm-1(弱,环内振动) 。对比实验和理论拉曼吸收光谱,它们的拉曼吸收峰基本是吻合的。

图3 模拟萘夫西林分子拉曼吸收光谱Fig.3 Laman of nafcillin
2.4 紫外吸收光谱
采用 TD DFT/B3LYP/6-31+G(d)理论方法, 模拟显示了萘夫西林分子在 247 nm (强吸收)处有强的紫外吸收峰,这是源于电子从最高占据轨道(HOMO)跃迁到最低空轨道(LUMO)所显示的吸收峰,在367 nm(弱吸收)处有弱的紫外吸收峰,这是源于电子从次高占据轨道(HOMO-1)跃迁到最低空轨道(LUMO)所显示的峰。这与其标准图谱显示萘夫西林分子在247.5 nm和357.5 nm 处有较强的紫外吸收峰基本是相吻合(见图4)。
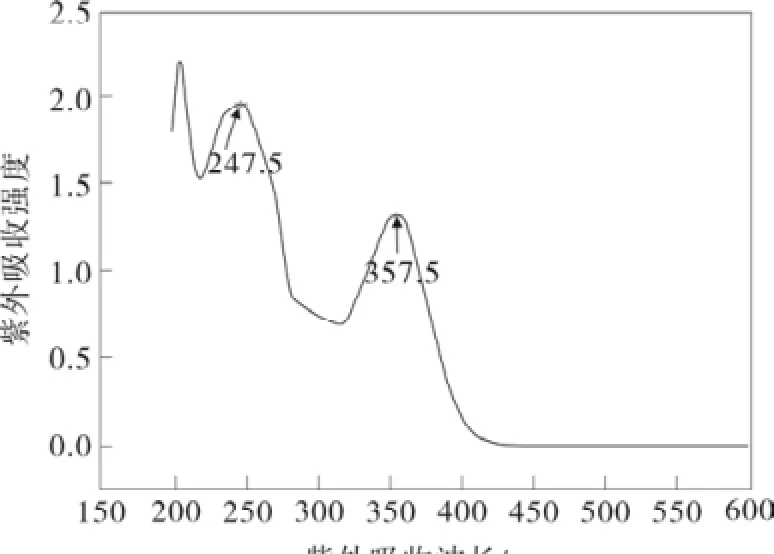
图4 萘夫西林分子紫外-可见吸收光谱Fig.4 UV-Vis of nafcilin
2.5 核磁共振光谱
用TMS B3LYP/6-311+G(2d,p) GIAO的方法理论模拟了萘夫西林分子的氢核磁共振谱(见图5所示)。
其萘环上六个氢(弱,7.218 9×10-6),乙氧基上的CH2(弱,3.581 4, 3.917 9×10-6); CH3(中,1.328 5,1.356 7,0.786 7×10-6); 酰胺键上的氢(弱,5.085 5 ×10-6) ,稠杂环6号位上的H(弱,4.216 9×10-6),5号位上的H(5.267 5×10-6),与3号位相连的2个CH3(强,1.521 3,1.291 1 ,1.199 7,1.292 8,1.269 7,1.157 8×10-6)。这些吸收峰较好地与其核磁共振实验吸收峰值吻合起来,验证了模拟计算的准确性[6-7]。

图5 萘夫西林的核磁共振氢谱Fig.5 1-HNMR of nafcilline
2.6 偶极矩和原子的自然电荷
分子的偶极矩是衡量其分子极性的重要物理量。采用 Gaussian 09W,我们计算得到标题分子的偶极矩为5.5418 Debye,这表明萘夫西林的极性比较大,血药浓度也较低,通过肌肉注射萘夫西林的血药浓度高于口服,但是仍然比较低。血浆蛋白结合率高,胆汁中的药的浓度高于血药浓度,所以尿液排泄量少,主要经胆汁排泄排出。从文献上查到的疏水参数计算参考值(logP)为2.9,这可能是因为萘夫西林的羧基的存在,增大了萘夫西林的亲水性和溶解度。
原子的自然电荷值是研究其化学活性点位及确定亲核或亲电特性强弱的一种有效方法。采用B3LYP/6-31+G(d)基组,我们分析了萘夫西林分子的自然键轨道值(NBO)。结果显示电荷最正的是羧基上的碳原子(8C,0.831e);其次为β-内酰胺环上的羰基上的碳原子(4C,0.711e);第三为酰胺键上的碳原子(15C,0.703e);第四为酰胺键上的氢原子(20H,0.433e),电荷最负的为羧基上的羟基氧(14O,-0.708e),其次为酰胺键上的氮原子(10N,-0.697e);第三为酰胺键上的氧原子(16O,-0.687e);第四为β-内酰胺环上的羰基上的氧原子(9O,-0.686e)。所以β-内酰胺环及与其相连的酰胺键(-CO-NH-)上的原子很可能是其发挥药理和药理活性的亲电和亲核反应中心。这也符合了青霉素类抗生素的药理是由于β-内酰胺类作用于细菌的细胞壁而产生的。
3 结 论
对萘夫西林分子((2S,5R,6R)-6-(2-乙氧基-1-萘酰氨基)-3,3-二甲基-7-羰基-4-硫杂-1-氮杂-二环[3.2.0]庚烷-2-羧酸),我们基于DFT理论和方法,在 6-31+G(d)基组水平上进行了分子结构优化。对其药物分子光谱(UV-Vis,IR, 1HNMR )等理化性质进行了理论模拟和指认,取得了与实验光谱基本吻合的结果,表明了本研究方法是可信的。模拟的几何优化结构表明, 萘夫西林分子具有稳定的分子构型和良好的疏水参数,在极性溶液中溶解能力将会较强。自然电荷(NBO)值计算结果表明,β-内酰胺环及与其相连的酰胺键(-CO-NH-)上的N原子很可能是其发挥药理和药理活性的亲电和亲核反应中心,符合青霉素类抗生素的药理是由于β-内酰胺类作用于细菌的细胞壁而产生。对其分子的模拟光谱及其指认,将有助于萘夫西林类药物的分析与检测。
[1]昝林森, 邱怀, 袁志发. 环核苷酸(CNT)在豚鼠体内代谢规律的研究[J]. 畜牧兽医学报,2000,12(2):39-45.
[2]张坐省, 傅建熙. β—受体兴奋剂在畜牧业生产上的意义[J]. 畜牧兽医杂志, 1996,14(4):34-38.
[3]涂秀英, 曹智勇, 聂复礼. 绞股蓝抗衰老药理实验研究[J]. 江西中医学院学报, 1999, 25(2):12-16.
[4]曾淑君,沈宝莲,文良珍. 云芝糖肽对裸鼠人肺腺癌抗癌作用的研究[J]. 中国药理学通报, 1995, 3(12):12-15.
[5]李敬芝, 姚锋. 老药新用[J]. 中国医刊, 1980, 12(1):22-25.
[6]彭聚宝.急性痛风的非秋水仙碱治疗[J]. 中国医院药学杂志, 1992, 31(2):21-24.
[7]黄庆国, 韩朔睽, 王连生. 利用前线分子轨道能预测有机物生物毒性的方法[J].环境化学, 1994, 12(1):23-26.
Density Functional Theory Study on Pharmacological Properties of Nafcilline
XV Yang,LUO Hai-xia,FAN Qiang,ZHONG Ai-guo
(College of Chemical Engineering and Pharmacy, Taizhou University, Zhejiang Taizhou 316000,China)
Using the density functional theory B3LYP method with 6-31+G(d) basis set,nafcilline molecules were optimized, and their molecular configuration, molecular dipole moment, IR spectra, UV spectra, NBO charge distribution were analyzed. The results show that, the maximum possible parts of pharmacological and toxicological effects of nafcilline are N atoms on the amide.
Nafcilline; Density functional theory; Quantitative calculation
TQ 624.2
: A
: 1671-0460(2015)04-0699-03
浙江省大学生科技创新项目(新苗人才计划),项目号:2014R428015。
2015-03-15
徐洋(1991-),男,浙江绍兴人,台州学院高分子专业,研究方向:从事高分子研究工作。E-mail:451872652@qq.com。
