抗癌药紫杉醇及其衍生物多西他赛药理及毒理性质的密度泛函研究
2015-01-10占凯峰裴诗恩钟爱国刘恩榕台州学院医药化工学院浙江台州318000
占凯峰,裴诗恩,钟爱国,刘恩榕(台州学院医药化工学院, 浙江 台州 318000)
抗癌药紫杉醇及其衍生物多西他赛药理及毒理性质的密度泛函研究
占凯峰,裴诗恩,钟爱国,刘恩榕
(台州学院医药化工学院, 浙江 台州 318000)
采用密度泛函理论B3LYP方法在STO-3G基组上对抗癌药紫杉醇和多西紫杉醇分子进行优化并进行理论计算,对它们的分子构型、偶极距、红外光谱(IR)、紫外光谱(UV-Vis)、NBO电荷分布进行了分析。结果表明,紫杉醇和多西紫杉醇发挥其药理和毒理作用的最大可能部位在酰胺基的N原子上。
紫杉醇;多西紫杉醇;密度泛函;量化计算
紫杉醇(taxel)是从红豆杉的树皮,种子,树叶和树根中提取出的以微管为作用靶点的抗肿瘤药物[1]。其主要药理作用为通过加快微管双聚体装配成微管,从而阻止细胞有丝分裂中去多聚化而使微管稳定,阻碍细胞有丝分裂于G2和M期,达到抑制癌细胞的分裂增殖的目的。紫杉醇在20世纪60年代被美国癌症研究中心发现于太平洋红豆杉的树皮中。自20世纪70年代发现其抗癌活性以来, 以各种方法合成了数百种类似物, 期望获得一种低毒副作用高生物活性的相似物, 但所得到的理想产物并不多。多西紫杉醇 (多西他赛,docetaxel) 是由法国一家公司开发的一种半合成紫杉醇衍生物。多西他塞对微管结合部位的作用效果是紫杉醇的2倍;当用来稳定微管和促进装配时,其活性是紫杉醇的3倍;当用来抑制微管解聚时,活性要是紫杉醇大2倍,在抗癌活性的体外效果试验中,已证实了多西他塞的效果是紫杉醇的1.3~12倍[2]。在理论计算方面,吴洪等探讨了电子结构的定量构效与紫杉醇抗癌活性的关系[3]。邱晓航等研究了紫杉醇及修饰物的光谱[4]。许旋等研究了紫杉醇的分子几何构型的从头计算及其核磁共振谱氢谱[5]。
药物为什么会有活性, 药物的化学结构与活性存在什么样的关系? 是人们一直在探索的重要问题。研究这些从实践中提出的问题,有助于认识药物与机体的作用规律。药物从给药到产生活性是一个非常复杂的过程,它涉及到药物在体内的吸收、分布、代谢和消除,又涉及到药物与生物靶点相互作用(阻断或刺激)的能力。近年来的研究表明,紫杉醇和多西他塞对免疫细胞具有特殊的调控作用,能产生类似于脂多糖所引起的天然免疫和特异性免疫, 对比直接作用于肿瘤细胞,它们作为免疫剂具有高药理活性低毒副作用等优点。通过密度泛函理论,对紫杉醇及多西他塞分子进行了计算,基于量子化学计算结果对它们作用机理进行初步探讨。
1 实验部分
对紫杉醇及多西紫杉醇(见图1)采用DFT理论的B3LYP方法在STO-3G基组水平上进行了优化和计算。优化得到的稳定构型基础上,采用Freq方法进行了频率分析, 结果表明所有简谐振动频率全部为正值, 表明其计算结果是可信的。本计算工作全部通过Gaussian09[6]程序包在PC机上完成。

图1 紫杉醇(上)和多西紫杉醇(下)结构和编号Fig.1 Molecular structure and atomic number of taxol(top) and docetaxel(down)
2 结果与讨论
2.1 分子几何构型分析
利用Gaussian 09 W程序包优化计算得到了紫杉醇分子的主要几何结构参数,包括键长,键角。正常的 C-N单键(0.148 nm),C-C单键(0.154 nm), C-O单键(0.143 nm), C=O双键(0.120 nm), O-H(0.098 nm),C=C双键(0.134 nm)。在共轭效应,诱导效应,空间效应作用下,键长会发生一定变化,优化后得到的表1中数据(略)基本在正常范围之内;结合分子结构分析,对应的双键长度要小于单键长度,符合稳定空间结构的构想要求。
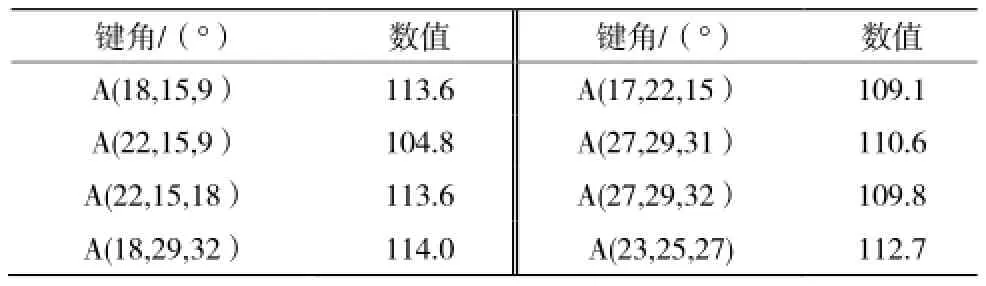
表1 优化后得到的多西紫杉醇的重要键角Table 1 The important bond angels optimized
从表1的数据中可以得知多西紫杉醇除苯环外的环中大多数键角均在109°至120°之间,没有发生明显的扭曲变形,而个别原子受空间位阻效应,邻位取代基效应影响比较大,键角发生了小幅度的变化,但这些都是在正常范围之内的,说明该结构是较为稳定的。对比多西紫杉醇 A(18,15,9)为113.6°,紫杉醇A(18,15,9)为115.6°,这可能是邻位取代基效应造成的。优化后的紫杉醇与多西紫杉醇的空间结构如图2所示。我们从图中可以看出,紫杉醇和多西紫杉醇都是较为复杂的空间分子,分子中的支链与环和主链有明显的扭转而没有处在同一平面,这样的结构受位阻效应小有利于发挥官能团的优势作用。紫杉醇和多西紫杉醇结构上主要的差别在于两个取代基的差异,紫杉醇 O22上连接的是羟基氧,而多西紫杉醇相同位置连接的是乙酰基;紫杉醇酰胺N上连接的是苯甲酰基,多西紫杉醇对应位置上连接的是烷氧基。多西紫杉醇的取代基空间位阻小,取代基极性基团亲水性大,由此推测多西紫杉醇的生物活性要比紫杉醇大。
2.2 NBO电荷分析
原子的 NBO电荷是研究分子的化学反应活性区域的亲核或者亲电特性强弱的有效方法。优化后的紫杉醇分子和多西紫杉醇各主要原子的 NBO电荷分布。由于N和O原子的电负性比C原子和H原子都要大,因此紫杉醇分子中的N(-0.342 e)和O(-0.167 ~ -0.307 e)原子都带一定的净负电荷,直接与N原子或者0原子相连的C原子都带一定的正电荷,这是因为与它们相连的O原子或者N原子强烈的吸引C原子或者H原子的电荷而使电子云向自身原子核偏移,导致自身电子云密度增加,C原子或H原子电子云密度降低;其他的C原子因为吸引氢原子上的电子云而都带一定的负电荷;对于氢原子而言,自身的电子都是被吸引偏离原子核而使电子云密度降低从而视偏移量都带一定量的净正电荷,一些带了大量正电荷的 H可能容易与其他物质形成氢键,可能是其发挥生物活性原因。对于连接在同一碳原子上氧,羰基氧的净负电荷要比单键氧的净负电荷要多,主要是因为羰基氧吸电子能力强,自身的电子云密度大。在由电荷控制反应中, 原子的负电荷越多, 其受亲电试剂进攻的可能性越大;反之, 原子正电荷越多, 则受亲核试剂进攻的可能性也越大。因此我们推测,紫杉醇中酰胺(N=-0.342 e),多西紫杉醇中酰胺(N=-0.352 e)为它们的主要亲电反应位置。
2.3 极性和疏水性
偶极矩和疏水参数是分子极性的关键物理量,也是表示药物亲脂性或疏水性的重要物理参数。通过使用 Gaussian 09程序包计算获得了紫杉醇的偶极距=6.196 5 D,多西紫杉醇的偶极距=4.821 3 D,说明紫杉醇的极性要大于多西紫杉醇。而紫杉醇与多西紫杉醇的疏水参数都比较大,这可能是由于紫杉醇与多西紫杉醇中苯环与大量甲基的存在,增大了它们的亲脂性的缘故。但多西紫杉醇的疏水参数要小一点,可能是多西紫杉醇要比紫杉醇多一个极性亲水基团羟基,少一个亲脂基团苯环的缘故,这将更有利于在人体内发挥其药理活性。
2.4 红外光谱
IR是研究分子的结构、化学键、表征和鉴别物质十分重要方法。在紫杉醇及多西紫杉醇结构优化基础上,模拟了紫杉醇及多西紫杉醇的红外振动光谱。紫杉醇属于C1群,有113个原子,对应333个振动模式。理论计算所得到的部分特征振动频率(0~4 000 cm-1)及红外线强度列于图2-上中,出现了紫杉醇特征峰值: 3 728 cm-1(O-H,伸缩), 3 296~3 375 cm-1(氢键O-H,伸缩), 1 623 cm-1(N-H,弯曲), 3 224 cm-1(N-H,伸缩),1 031~1 383 cm-1(C-O,伸缩),1 806~1 825 cm-1(C=O,伸缩),1 353 cm-1(酰胺C-N,伸缩),1 238 cm-1(胺C-N,伸缩),1 789 cm-1(C=C,伸缩),1 584~1 613 cm-1(苯环面内伸缩),660~710 cm-1(苯环骨架面外变形振动)1 065 cm-1(苯环呼吸);多西紫杉醇(见图2-下)属于C1群, 有111个原子,对应327个振动模式,多西紫杉醇的特征峰:3 733 cm-1(O-H,伸缩), 3 293~3 359 cm-1(氢键O-H,伸缩), 1 622 cm-1(N-H,弯曲), 3 194 cm-1(N-H,伸缩),1 029~1 381 cm-1(C-O,伸缩),1 806~1 855 cm-1(C=O,伸缩),1 353 cm-1(酰胺C-N,伸缩),1 238 cm-1(胺C-N,伸缩),1 795 cm-1(C=C,伸缩),1 584~1 613 cm-1(苯环面内伸缩),660~710 cm-1(苯环骨架面外变形振动),1 065 cm-1(苯环呼吸); 红外光谱的特征峰特点较好地与官能团吻合起来,也进一步验证了计算的准确性。

图2 紫杉醇(上)和多西紫杉醇(下)红外谱图Fig.2 IR Spectra of taxol (top) and docetaxel (bottom)
2.5 核磁共振谱
核磁共振谱1H-NMR和13C-NMR能反映化合物的结构,1H-NMR主要根据测定有机物分子中氢原子的化学环境,即反应分子的空间构像来推测有机物的结构。在紫杉醇的结构优化基础上,在HF/6-31G水平上计算模拟了紫杉醇的H原子的核磁共振图谱(图3),并列出部分H原子的模拟计算值与实验值对比于表。为了更能直观地反应1H-NMR计算值与实验值的符合程度,以1H-NMR计算值为纵坐标,以1H-NMR实验值为横坐标做散点图。可以观察到各点较均匀分布在拟合直线的两侧,这说明这次计算是较为可信的。
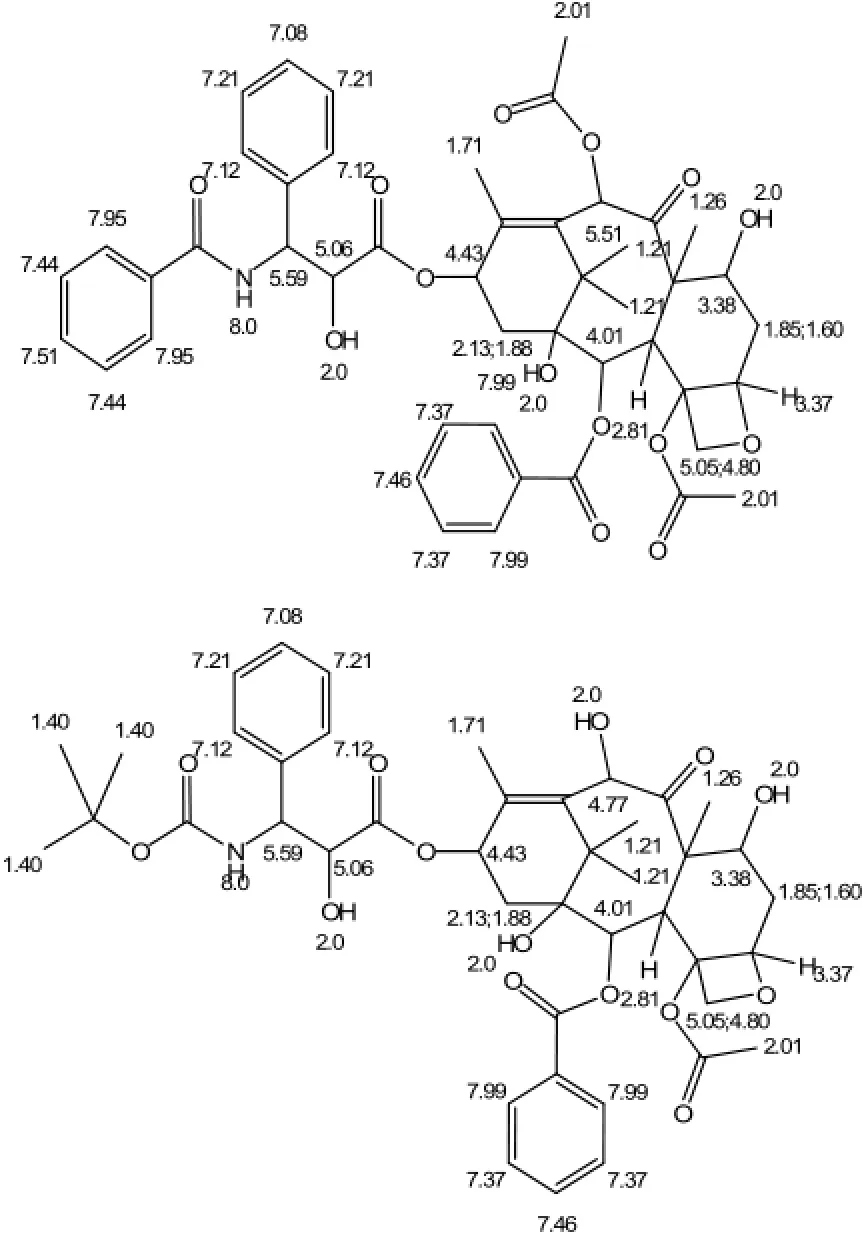
图3 紫杉醇(上 )和多西紫杉醇(下)HNMR图谱Fig.3 The HNMR spectra of taxol (top) and docetaxel (bottom)
3 结 论
通过对紫杉醇分子和多西紫杉醇分子的密度泛函量化计算研究,得到以下结论并作了一些推测:
(1)紫杉醇分子和多西紫杉醇分子具有稳定的分子结构,易溶于丙酮氯仿等有机溶剂,在水中的溶解度都很小,但多西紫杉醇的溶解度要稍大于紫杉醇,红外光谱与官能团大致符合,核磁共振计算值与实验值较为符合。
(2)通过NBO电荷分布分析,推测紫杉醇分子与多西紫杉醇分子活性最强的部位在酰胺基的N上,并且多西紫杉醇分子的活性要比紫杉醇分子强。
[1]陈晓萍, 薛兴奎, 张沂平, 等. 多烯紫杉醇对胃癌细胞作用及其机制的研究[J]. 肿瘤, 2006, 26(4): 343-345.
[2]姚和际. 多西紫杉醇研究进展及市场分析[J]. 中国制药信,2001, 17(5): 29-30.
[3]吴洪, 许锦泉, 谢少齐, 等. 紫杉醇抗癌活性与电子结构的定量构效关系[J]. 泉州泉州师范学院学报,2008,26(2):63-66.
[4]邱晓航, 欧阳砥, 巢晖, 周永洽. 紫杉醇及其修饰物的光谱研究[J].高等学校化学学报,2000,21(3):415-416.
[5]许旋,徐志广,罗一帆. 紫杉醇的核磁共振谱及其分子几何构型的从头计算[J]. 物理化学学报,2002,18(5):420-425.
[6]M. J. Frisch, G. W. Trucks, H. B. Schlegel,et al. Gaussian 09, Revision C.02[R]. Gaussian, Inc., Wallingford CT, 2009.
Density Functional Theory Study on Pharmacology and Toxicology of Taxol and Its Derivative Docetaxel
ZHAN Kai-feng,PEI Shi-en,ZHONG Ai-guo,LIUEn-rong
(College of Chemical Engineering and Pharmacy, Taizhou University, Zhejiang Taizhou 318000,China)
Using the density functional theory B3LYP method with STO-3G basis set, molecules of taxol and docetaxel were optimized, and the theoretical calculation was carried out; their molecular dipole moment and configuration, IR spectra, UV spectra, NBO charge distribution were analyzed. The results show that, the maximum possible parts of pharmacological and toxicological effects of taxol and docetaxel are their N atoms on the amide.
Taxol; Docetaxel; Density functional theory; Quantitative calculation
TQ 623.4
: A
: 1671-0460(2015)04-0684-03
浙江省大学生科技创新项目(新苗人才计划),项目号:2014R428015。
2015-03-15
占凯峰(1993-),男,浙江杭州人,台州学院高分子专业,研究方向:从事高分子研究工作。E-mail:670478214@qq.com。
