天然产物(+)-Balasubramide 的合成
2015-01-08易鑫宇林汉森
李 骏,易鑫宇,定 力,林汉森
广东药学院药科学院,广州 510006
(+)-Balasubramide 是从芸香科黄皮属植物山黄皮叶[Clausena indica (Datz).Oliv.]的氯仿提取物中分离出来的一种八元内酰胺类化合物[1],其绝对构型为5S,6R[2](图1)。同时被分离出来的还有其生物合成前体(+)-prebalamide(图1)。研究表明,从黄皮叶中分离出来的其它内酰胺类化合物,如五元内酰胺类化合物黄皮酰胺(图1),具有肝细胞保护作用[3-5]、神经保护作用[6,7]、抗细胞凋亡作用[8,9]、抑制细胞脂质过氧化和清除氧自由基作用[10,11]等多种药理活性,而八元内酰胺类化合物的生物活性却鲜有报道。作为新型先导物的(+)-balasubramide,其结构虽较为新颖,但由于其八元内酰胺的特殊结构以及含有两个手性中心,使其全合成有一定困难,因此,寻找一条简单、高效的合成路线,对化合物进行更进一步生物活性研究具有重要意义。

图1 (+)-Balasubramide、(+)-prebalamide 和(±)-黄皮酰胺的结构Fig.1 Chemical structure of (+)-balasubramide,(+)-prebalamide and (±)-clausenamide
目前,关于(-)-balasubramide 的合成有3 种方法[12-14],但对于天然产物(+)-balasubramide 的合成方法却仅只有1 种。2007 年,Johansen 等人[13]通过对中间体(±)-β-苯基-α,β-环氧丙酸钾进行手性拆分,首次完成了(+)-balasubramide 及其对映体的合成,共四步反应,但由于拆分环节将损失一半的对映体,故拆分方法总收率仅为17%,ee 值为98%。
此前,本课题组已发现,以肉桂醛为原料,使用仲胺类化合物S-二苯基脯氨醇三乙基硅醚(4)为手性催化剂,一锅法不对称合成(2S,3R)-(+)-β-苯基-缩水甘油酸甲酯(1)[15],合成路线如图2。

图2 (2S,3R)-(+)-β-苯基缩水甘油酸甲酯的不对称合成Fig.2 Asymmetric synthesis of methyl (2S,3R)-3-phenyloxirane-2-carboxylate
以此为基础,本文继续对化合物1 的合成工艺条件进行优化,从而有效提高制备的总产率,然后以其为起始底物,经酯胺缩合反应和分子内环合反应,得到天然产物(+)-balasubramide,三步反应总收率为44%(从肉桂醛算起),ee 值达到>99%。其结构经1H NMR、13C NMR、HR-MS 和IR 确证。合成路线如图3。

图3 天然产物(+)-balasubramide 的不对称全合成Fig.3 Total asymmetric synthesis of natural product (+)-balasubramide
试剂和条件:(a)手性催化剂,30%过氧化氢溶液,N-溴代丁二酰亚胺,无水碳酸钠;(b)N-甲基色胺,甲醇钠,甲醇,-18 ℃,2 d;(c)三氟甲磺酸化镱,四氢呋喃,室温
Reagents and conditions:(a)chiral catalyst,30%H2O2,NBS,anhydrous Na2CO3;(b) N-Methyltryptamine,CH3ONa,CH3OH,-18 ℃,2 d;(c)Yb(CF3SO3)3,THF,rt
1 材料与方法
1.1 仪器与试剂
1H NMR 谱和13C NMR 采用Bruker Advance 500型核磁共振仪(500 MHz,CDCl3或DMSO-d6为溶剂);质谱采用Waters 液相色谱质谱联用仪(ESITOF);红外测试采用德国Bruker-EQUINX55 型傅里叶变换红外光谱仪;熔点测定使用北京泰克仪器有限公司X-5 型熔点测定仪,温度未经校正;旋光仪使用WXG-4 型圆盘旋光仪;高效液相采用Agilent 1200;手性柱使用Daicel Chiralcel AD-H(4.6 mm ×25 cm);柱色谱硅胶(青岛海洋化工);实验使用试剂均为市售分析纯或化学纯。
1.2 (+)-balasubramide 的合成
1.2.1 (2S,3R)-(+)-β-苯基-缩水甘油酸甲酯(1)的合成
称取肉桂醛3.776 g(30 mmol),室温下加入到30% H2O23.677 mL(36 mmol)和手性催化剂1.103 g(3 mmol)的25 mL 二氯甲烷溶液中,室温下反应2 h 后,依次加入无水Na2CO33.816 g(36 mmol),甲醇30 mL 和NBS 6.408 g(36 mmol),加毕,室温下反应3 h(TLC 检测)。反应结束后,过滤,滤渣用二氯甲烷(15 mL ×3)洗涤,合并滤液洗液,用水洗(50 mL×2),有机层用无水硫酸钠干燥后,浓缩后得橙色油状物,柱层析纯化(洗脱剂:石油醚:乙酸乙酯=10:1),得浅黄色油状物4.6 g,产率87%。ee 值为95%(n-hexane/i-PrOH=90/10,λ=220 nm,0.8 mL/min,tmajor=18.4 min,tminor=19.7 min);=+156.3 (c=1.3,CHCl3);1H NMR (500 MHz,CDCl3)δ 7.40~7.34 (m,3H),7.32~7.28 (m,2H),4.11 (t,J=2.7 Hz,1H),3.83 (s,3H),3.53(d,J=1.8 Hz,1H)。
1.2.2 N-甲基-N-(乙基-2-(1H-吲哚基))-3-苯基环氧乙烷基-2-胺(2)的合成
将N-甲基色胺(1.74 g,10 mmol)溶于20 mL 甲醇中,干冰浴冷却到-18 ℃,然后滴加MeONa(0.54 g,10 mmol)甲醇溶液10 mL,滴加完毕后加入化合物1(2.14 g,12 mmol),混合均匀,在-18 ℃冰箱放置2 d,TLC 检测,反应结束后,于冰浴下加入10 mL的蒸馏水,用0.2 N 的盐酸溶液调节pH 至8~9,用二氯甲烷(20 mL ×5)萃取,合并有机层,并加入无水硫酸钠干燥,浓缩,得到黄色油状物,产品不需要纯化直接进行下步反应。
1.2.3 (5S,6R)-(+)-3-甲基-5-羟基-6-苯基-1,2,3,5,6,7-六氢-4H-吖辛因[5,4-b]吲哚-4-酮(3)的合成
将化合物2(0.70 g,2.19 mmol)溶于30 mL 四氢呋喃中,然后加入三氟甲磺酸化镱(271.56 mg,0.438 mmol)。室温反应,TLC 检测,反应结束后减压蒸除溶剂,残渣加入50 mL 二氯甲烷溶解,然后用饱和NaCl 溶液(10 mL × 3)洗涤,水洗,无水硫酸钠干燥,减压蒸除溶剂,得粘稠物,粗品经柱层析纯化(洗脱剂:石油醚-乙酸乙酯=3∶2),再经真空干燥,得白色固体0.57 g,产率50%(从化合物1 算起)。ee 值为>99%(n-hexane/i-PrOH=40/10,λ=254nm,0.8 mL/min,tmajor=18.5 min,tminor=33.2 min);mp.64~66 ℃;[α]16
D=+4.0 (c=0.5,MeOH);1H NMR (500 MHz,DMSO)δ 10.77 (s,1H),7.50 (d,J=7.8 Hz,1H),7.34-7.27 (m,4H),7.23~7.16 (m,2H),7.03-6.93 (m,2H),5.22 (d,J=8.3 Hz,1H),4.98 (t,J=8.5 Hz,1H),4.27 (d,J=8.6 Hz,1H),4.12 (dt,J=15.1,7.7 Hz,1H),3.57~3.48 (m,1H),3.43 (dt,J=16.0,8.0 Hz,1H),3.04 (ddd,J=16.0,7.3,4.6 Hz,1H),2.59 (s,3H);13C NMR (126 MHz,DMSO)δ 174.26,142.69,135.50,134.75,129.55,128.69,128.61,126.96,121.34,118.77,117.89,110.96,105.56,79.66,52.61,46.40,34.52,22.50;IR(KBr,cm-1):3455,3245,2361,1651,1624,1496,1459,1072,738,699,504;HRMS (ESI)caculated for C20H21O2N2[M+H]+=321.1603;found:321.1600。
2 结果与讨论
2.1 一锅法化合物1 的合成
2.1.1 氧化酯化中投料顺序对反应收率的影响
按文献[15]方法制备化合物1,收率为72%,后经多次考察,我们发现投料顺序的不同,对产物最后的收率影响较大,投料顺序对反应收率的影响,见表1。由表1 可见,依次按照无水碳酸钠、甲醇、NBS 的顺序投料,反应的收率最高。从氧化酯化的机理分析,无水碳酸钠的率先加入,有效吸收不对称环氧化中过氧化氢产生的水,从而推动反应向右进行,很好的提高收率,而甲醇与NBS 的加入顺序则对收率影响不大。

表1 氧化酯化的投料顺序优化Table 1 Optimal feeding sequence of oxidative esterification
2.2 酯胺缩合反应
以甲醇为溶剂,在室温条件下,将化合物1 与N-甲基色胺发生酯胺交换反应,放置两天,但仍未见产物生成。随后,尝试在低温条件下,加入催化量的碱如NaHCO3、K2CO3、Na2CO3、t-BuOK 和MeONa。结果发现以t-BuOK 为碱时,检测到有产物生成,但副产物过多,而以MeONa 为碱时则最佳,可获得较好的产率。其可能的反应机理如图4。这里选用甲醇钠为碱催化剂,可防止其他亲核试剂对化合物1羰基进行进攻的干扰,有效减少副反应的发生。此外,由于甲氧负离子与N-甲基色胺相比,位阻较小且亲核性较强,所以在两者的亲核取代竞争中,甲氧负离子占有绝对优势,而该反应能够进行的原因是3-(2-氨乙基)吲哚基的离去性较差。因此,酯胺缩合反应的速率较慢(需要2 d)。在柱层析纯化时,产物斑点附近总伴随一个微弱的未知杂质点,粗产物未能得到绝对的纯化,推测化合物在柱层析分离过程中不稳定而产生分解,但产物可不经纯化而进入下一步反应。
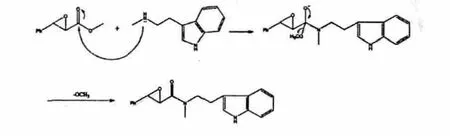
图4 化合物1 酯胺交换可能的反应机理Fig.4 Proposed mechanism for amine-ester interchange of compound 1
2.3 分子内环合反应
链状酰胺(2)进行分子内环合时,我们发现选用活性不同的路易斯酸做催化剂,对环合反应影响很大。如用AlCl3、FeCl3或CuCl 催化时,反应没有目标产物生成,当用LaCl3、p-TSA 或Yb(OTf)3催化时,检测到目标产物的生成,其中以三氟甲磺酸化镱(Yb(OTf)3)最佳,环合反应可获得73%的收率和>99%的ee 值。而溶剂对环合反应产物的ee 值没有影响,但对环合产率有一定影响,其中以极性溶剂四氢呋喃最佳,收率达82%。对Yb(OTf)3催化链状酰胺(2)进行分子内环合的机理推断如图5,Yb(OTf)3与环氧键上的氧原子进行配位结合,发生了类似SN2 机理的取代反应。
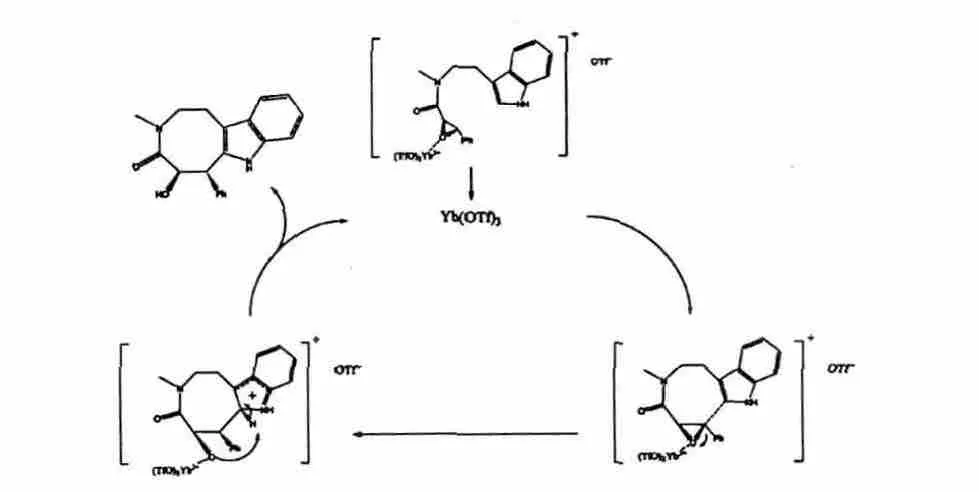
图5 化合物2 分子内环合可能的反应机理Fig.5 Proposed mechanism for intramolecular cyclization of compound 2
3 结论
本文阐述了一条简单、温和、高效的天然产物(+)-balasubramide 的合成方法,以肉桂醛为起始原料,一锅法不对称合成(2S,3R)-(+)-β-苯基-缩水甘油酸甲酯,再经酯胺缩合和分子内环合共三步反应,不对称合成天然产物(+)-balasubramide,总收率为44%,ee 值为>99%。本合成方法化学收率和光学收率都较高,可为进一步研究该天然产物的生物活性及构效关系奠定一定基础。
1 Riemer B,Hofer O,Greger H.Tryptamine derived amides from clausena indica.Phytochemistry,1997,45:337-341.
2 Juárez-Calderón M,Aparicio DM,Gnecco D,et al.Synthesis of the indoloazocine derivatives from a chiral indol amide-stabilized sulfurylide.Tetrahedron Lett,2013,54:2729-2732.
3 Yang MH (杨明河),Cao YH (曹延怀),Li WX (李伟勋),et al.Isolation and structural elucidation of clausenamide from the leaves of Clausena lansium (lour.)skeels.Acta Pharm Sin (药学学报),1987,22:33-40.
4 Yang MH (杨明河),Chen YY (陈延镛),Huang L (黄量).Studies on the chemical constituents of Clausena lansium (lour.)skeels.Acta Chem Sin (化学学报),1987,45:1170-1184.
5 Yang MH,Chen YY,Huang L.Three novel cyclic amides from Clausena lansium.Phytochemistry,1988,27:445-450.
6 Xue W (薛薇),Zhang W (张威),Chen NH (陈乃宏).Advances in the study of chiral clausenamide.Chin J New Drugs (中国新药杂志),2008,17:268-271.
7 Zhang J (张静),Cheng Y (程勇),Zhang JT (张均田).Protective effect of (-)-clausenamide against neurotoxicity induced by okadaic acid and β-amyloid peptide25-35.Acta Pharm Sin (药学学报),2007,42:935-942.
8 Yao QQ (姚庆强),Wang Y (王琰),Yang SM (杨树民),et al.Biotransformation of (+)-and (-)-clausenamide in rats.Acta Pharm Sin (药学学报),2001,36:224-228.
9 Wang RS (王润生),Zhang JT (张均田).Construction of Bax α high expressing PC-12 cell line and the mechanisms of(-)-clausenamide in inhibiting apoptosis.Acta Pharm Sin(药学学报),2000,35:404-407.
10 Lin TJ (林童俊),Liu GT (刘耕陶),Li XJ (李小洁),et al.Anti-lipid peroxidation and oxygen free radical scavenging activity of clausenamide.Chin J Pharm Toxicol (中国药理学与毒理学杂志),1992,6:97-102.
11 Jiang XY,Zhang JT.Study on the noot ropic mechanism of(-)-clausenamide-influence on the formation of synapses in mouse brain.J Asian Nat Prod Res,1998,1:53-58.
12 Yang L,Deng G,Wang DX,et al.Highy efficient and stereoselective N-vinylation of Oxiranecarboxamides and unprecedented 8-endo-epoxy-arene cyclization:expedient and biomimetic synthesis of some clausena alkaloids.Org Lett,2007,9:1387-1390.
13 Johansen MB,Leduc AB,Kerr MA.Concise biomimetic total syntheses of both antipodes of balasubramide.Synlett,2007,16:2593-2595.
14 Zheng CW,Li YW,Yang YQ,et al.Highly efficient asymmetric epoxidation of electron-deficient α,β-enones and related applications to organic synthesis.Adv Synth Catal,2009,351:1685-1691.
15 Xuan YN,Lin HS,Yan M.Highly efficient asymmetric synthesis of α,β-epoxy esters via one-pot organocatalytic epoxidation and oxidative esterification.Org Biomol Chem,2013,11:1815-1817.
