从氨的合成认识氮分子化学键的稳定性和活化
2015-01-05杨奇谢钢陈三平
杨奇 谢钢 陈三平
(西北大学化学与材料科学学院 陕西西安 710079)
从氨的合成认识氮分子化学键的稳定性和活化
杨奇 谢钢 陈三平
(西北大学化学与材料科学学院 陕西西安 710079)
先从氮分子的化学键讨论其稳定性,再从最早工业化的合成氨出发,讨论氮分子化学键活化的相关研究,意欲探索一种元素化学新的教学方法。目的在于引导学生灵活学习并运用知识去认识自然世界,提高分析问题和解决问题的能力。
氨的合成 固氮酶 分子氮配合物 教学方法
作为自然界中氮单质最普遍形态的氮气在标准状况下是一种无色、无味、无臭的双原子气体分子。氮气占地球大气总体积的78.09%,是含量最多的气体[1]。工业上很多重要的化合物,如氨、硝酸、用作推进剂或炸药的有机硝酸盐以及氰化物都含有氮原子。
N2中的两个氮原子之间具有非常牢固的化学键,正因为此,无论是在工业中还是在生物体内,将N2转化为含氮化合物都很不容易。相应的,当含氮化合物燃烧、爆炸或分解时会产生氮气,通常释放大量的能量。如式(1)所示,联氨在空气中燃烧,产生大量的热并放出氮气。因此联氨及其衍生物可用作导弹及火箭的燃料。
(1)
同样,含氮化合物作为肥料和在能量储存方面的应用对人类非常重要。因此,研究氮气分子化学键的稳定性及其活化条件,利用空气中存在的取之不尽的氮气原料合成氨及含能化合物在人类经济活动中占有重要的地位,已成为化学工作者的热点课题之一。
本文先从N2分子的化学键讨论其稳定性,再从最早工业化的合成氨出发,讨论N2分子化学键活化的相关研究。目的在于引导学生如何灵活学习和运用知识去认识自然世界,提高分析问题和解决问题的能力。
1 氮分子的稳定性
氮分子(N2)中的三键(一个σ键和两个π键,图1)是最强的化学键之一,键能达946 kJ/mol[2]。这就是将N2转化为其他氮化合物非常困难的原因之一。
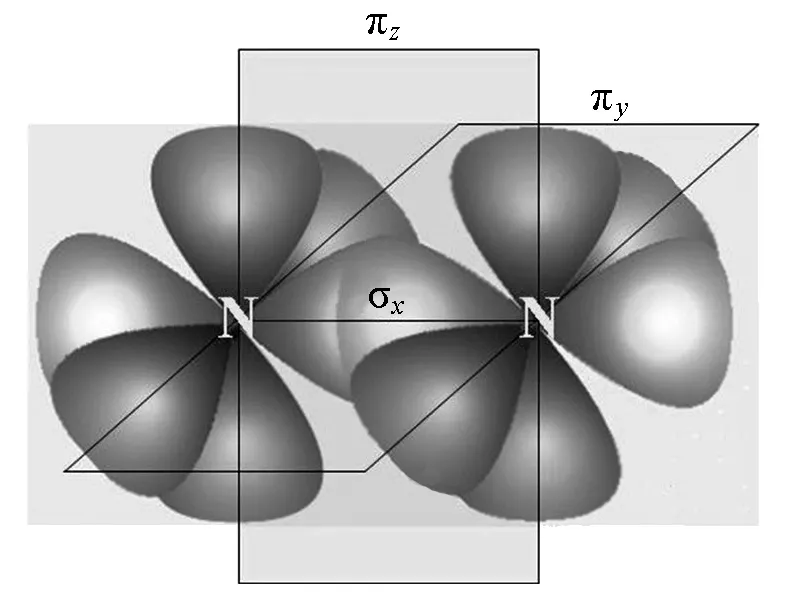
图1 氮分子的结构图
由紫外光电子能谱实验和理论计算可得到氮的分子轨道能级[3-5]。由N2分子基态的能级状态[6](图2)可见,N2分子基态的电子结构为:
KK(σg2s)2(σu2s)2(πu2p)4(σg2p)2
换句话说,N2分子的HOMO是σg2p,它的能量很低,N2的电离能(15.58 eV)接近氩(15.75 eV)。在N2分子中,πu2p分子轨道的能量比σg2p低,而电离能高;同时,N2分子的πg2p轨道(LUMO)比σg2p(HOMO)能量高8.6 eV,仅能由电正性高的碱金属等提供的电子所占领,而在σg2p及πg2p之间的间隙大,又无其他的轨道,使分子不易发生简单的电子转移氧化还原过程:
(2)
(3)
这就是N2分子不活泼性的原因之二。
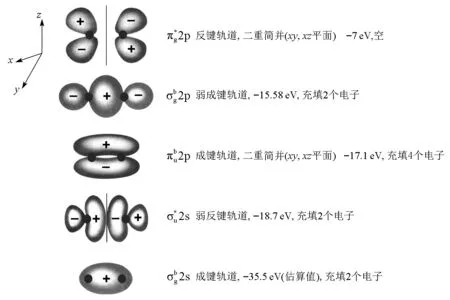
图2 N2分子轨道能级图
N2分子不活泼性的原因之三在于它的极化率很低(1.53 × 10-24cm3),难以形成亲电和亲核取代反应中常常涉及的那种高极性过渡态。
虽然N2分子在通常情况下是非常稳定的,但这种稳定又是相对的: ① 豆科植物根部可固氮; ② 金属锂在空气中可生成1∶2的Li2O和Li3N黑色壳; ③ 在雷电条件下,空气中的N2和O2会化合生成氮的氧化物; ④ 光催化合成,如式(4)所示:
2 氮分子的活化及固氮
将大气中游离N2转化为NH3的过程叫做氮的固定。在世界范围内,有一大批杰出科学家将注意力集中于这个问题,他们想方设法使稳定的氮分子的三键弱化并生成NH3,以满足人类的需求。
2.1 人工合成氨
世界粮食产量虽年年有所增长,但人口增长又造成粮食消费的快速增长。按照联合国人口基金会的预测,到2050年,世界人口将增至91.5亿。显然,激增的人口与粮食供求的矛盾愈发凸显。
德国化学家哈伯(F.Haber)从1902年开始研究由氮气和氢气直接合成氨。他于1908年申请了“循环法”专利,并在此基础上,于1909年又改进了合成方法,使氨的含量达到6%以上[7-9]:
该法在工业中普遍采用,即直接合成法。当今世界上有1/3的粮食产量直接来源于施用化学肥料所导致的增产。这意味着,如果沒有化肥工业,在20世纪,全世界有20亿人会因饥饿而丧生。化肥的合成结束了人类完全依靠天然氮肥的历史,将人类从饥饿中拯救了出来。哈伯因此获得1918年诺贝尔化学奖。
热力学计算表明,低温、高压有利于合成氨反应,但无催化剂时,反应的活化能很高(Ea= 420 kJ/mol)[10],反应几乎不发生。当采用铁催化剂时,由于改变了反应历程,降低了反应的活化能,使反应以显著的速率进行。后来,德国化学家G.Ertl阐明了合成氨反应过程由以下几个步骤(Haber-Bosch过程)构成[11]:
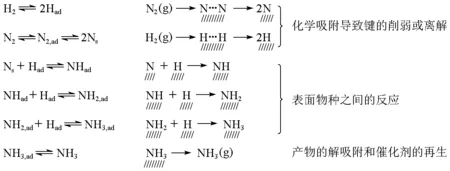
最后NH3离开催化剂表面。在图3中给出了每一步反应的势能[12]。2007年,G.Ertl由于对表面化学研究的贡献而获得诺贝尔化学奖[13]。
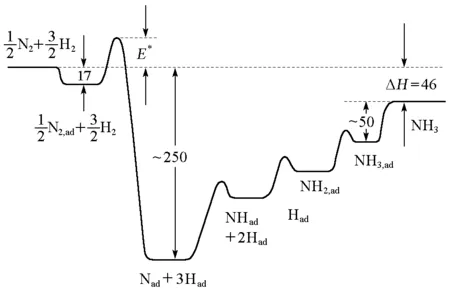
图3 合成氨反应势能图
直接合成法不但消耗能源,设备昂贵,而且还加重了大气污染和温室效应,破坏了生态平衡。因此,科学家也在寻找开发氨合成节能技术。2012年,东京工业大学细野秀雄研究小组报告说[14],他们以开发的超导物质C12A7(钙铝酸盐化合物,高铝水泥的主要成分)与钌微粒作用,制成催化剂C12A7:e-。研究发现,在该催化剂作用下,氮和氢能高效合成氨,消耗的能源仅为传统方法的十分之一。他们认为,这是由于在化合时相关电子变得容易移动,使氮分子容易成为原子。目前这种技术尚处在实验室阶段。
常温、常压条件的固氮问题一直在挑战着人类智慧,科学家在努力寻求建立温和条件下合成氨的新体系。真正常温、常压下固氮的研究起始于某些植物的根瘤菌能将大气中游离N2转化为NH3的事实。研究的一种思路是根据根瘤菌中固氮酶的组成、结构和固氮过程来模拟生物固氮;另一种思路是通过过渡金属的分子氮配合物活化N≡N键,进而通过适当的反应得到NH3。两种思路都是从如何削弱分子氮的三键入手的。
2.2 模拟生物固氮
本文无意详细阐述模拟生物固氮的研究进展,只简介几个相关的概念。有兴趣的读者可参看一些综述文章[15-19],从中获取更多的信息。
2.2.1 固氮酶[15]
固氮酶(nitrogenase)是某些微生物在常温常压下固氮成氨的主要催化剂,它能将生物体无法直接利用的分子氮(N2)转化成可利用的氨态氮(NH3),而且不需要如工业合成氨过程那样消耗大量的能源,不降低土壤活性,不污染环境。全球每年约有2.4亿吨的氨态氮是通过微生物的固氮过程实现的,约占全球氮资源的65%,该过程是自然界实现氮循环的重要环节[20]。固氮酶是由两种蛋白质组成的:一种含铁,称为铁蛋白;另一种含铁和钼,称为钼铁蛋白。所有固氮酶都具有一个含铁和硫的辅因子(包括活性区域中的一个四聚体,例如FeMoCo)。大多数固氮酶拥有一个中央钼原子,其他金属的则由钒原子或铁原子取代[21]。只有钼铁蛋白和铁蛋白同时存在,固氮酶才具有固氮的催化作用,因为这两种物质作为电子载体能够起到传递电子的作用。
能合成固氮酶的生物有两类: ① 非寄生固氮菌,如蓝藻(通过特异化的异形细胞)和固氮菌科的菌类; ② 共生固氮菌,如根瘤菌和弗兰克氏菌。
2.2.2 固氮酶的固氮机理[16-19]
在固氮酶中,固氮的总反应可以表示为[22]:
(6)
图4给出了固氮酶的固氮机理[23]:在ATP水解作用下,电子由还原剂传递给Fe蛋白,再由Fe蛋白传递给FeMo蛋白,在质子参与下,将N2还原为NH3,同时释放出H2。据推测,在FeMo蛋白中,电子先经过P簇(图5),再传递给活性中心FeMo辅基[24]。研究还发现,固氮酶中发生的是单电子传递,并且电子由Fe蛋白传递给FeMo蛋白是整个反应的决速步[25]。
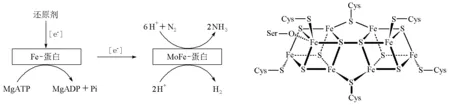
图4 固氮酶的固氮机理示意图 图5 P簇的结构示意图
固氮酶的固氮机理说明了两个问题:一是产生氢原子(这是由FeMo辅基以外的固氮酶各组成部分完成的,即铁蛋白的氧还循环);二是活化还原底物(这是由FeMo辅基完成的,即钼铁蛋白的氧还循环。当然,这必须要有前者存在)。这便是根据固氮酶反应动力学提出的Thorneley-Lowe模型[26],对人工模拟生物固氮很有启发意义。
FeMo辅基活化还原底物N2的作用方式已被广泛地探讨[23,27-33]。目前普遍认为FeMo辅基中所存在的6个不饱和配位的Fe是还原底物被催化活化的部位,并且一般都认为FeMo辅基与还原底物的作用方式随着还原底物的变化而变化。当还原底物的体积较小时,还原底物进入FeMo辅基的内部(通过由4个Fe和4个S所组成的“口腔”);当还原底物的体积较大时,还原底物只能在FeMo辅基的外部被活化。但对于固氮酶的天然底物N2的作用方式目前仍存在较大的争议。这是由于N2的体积与FeMo辅基的“口腔”相近,且略比FeMo辅基的“口腔”大一些(约大0.04 nm)。其中有代表性的FeMo辅基与N2的作用方式有3种[28,31-33](图6)。
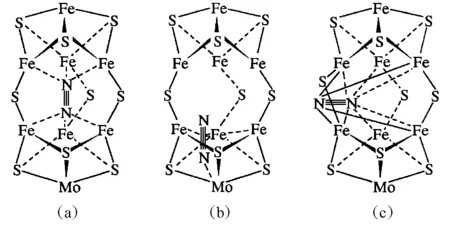
图6 FeMo辅基与N2的3种作用方式示意图
2.2.3 固氮酶的结构
生物固氮主要是通过固氮生物体内的固氮酶的催化作用完成的。如此讲来,弄清固氮酶的结构对于揭示固氮酶的固氮机理和人工模拟生物固氮研究至关重要。20世纪90年代以来, 通过固氮酶和辅因子的X射线衍射结晶学研究, 对于蛋白之间、亚基之间、蛋白与辅因子之间的相对位置和相互作用方式有了更为精确的认识。特别是固氮酶的活性中心原子簇及其周围蛋白质分子的三维结构的阐明, 是固氮酶结构研究的重大突破。
自从1977 年Shah 和Brill[34]分离出活性中心FeMo 辅基以来,无机化学家根据固氮酶中铁、钼、硫原子的比例(当时确定为7∶1∶8)并运用他们丰富的配位化学知识,曾经合成了多种模型化合物,其中某些化合物已逼近固氮酶的真实结构。然而,真正搞清其结构却经历了20年。固氮酶结构的确定使人们在分子-原子水平上对固氮酶的本质有了进一步的认识。
对固氮酶结构的研究经历了漫长的岁月。1960年,Camahan等[35-36]成功地建立了具有稳定固氮活性的无细胞提取物的方法;不久,Morteson等[37]首先确定了固氮酶是双组分酶的复合体;之后,Kennedy等[38]通过SDS-PAGE证明铁钼蛋白有两种亚基。固氮酶结构研究的3个里程碑是: ① 1992~1994年,Kim等发表的空心结构[39-42]; ② 2002年,由Morteson等提出的N为填隙原子的结构[43]; ③ 2011年,由Kennedy等提出的C为填隙原子的结构[44-45](图7)。
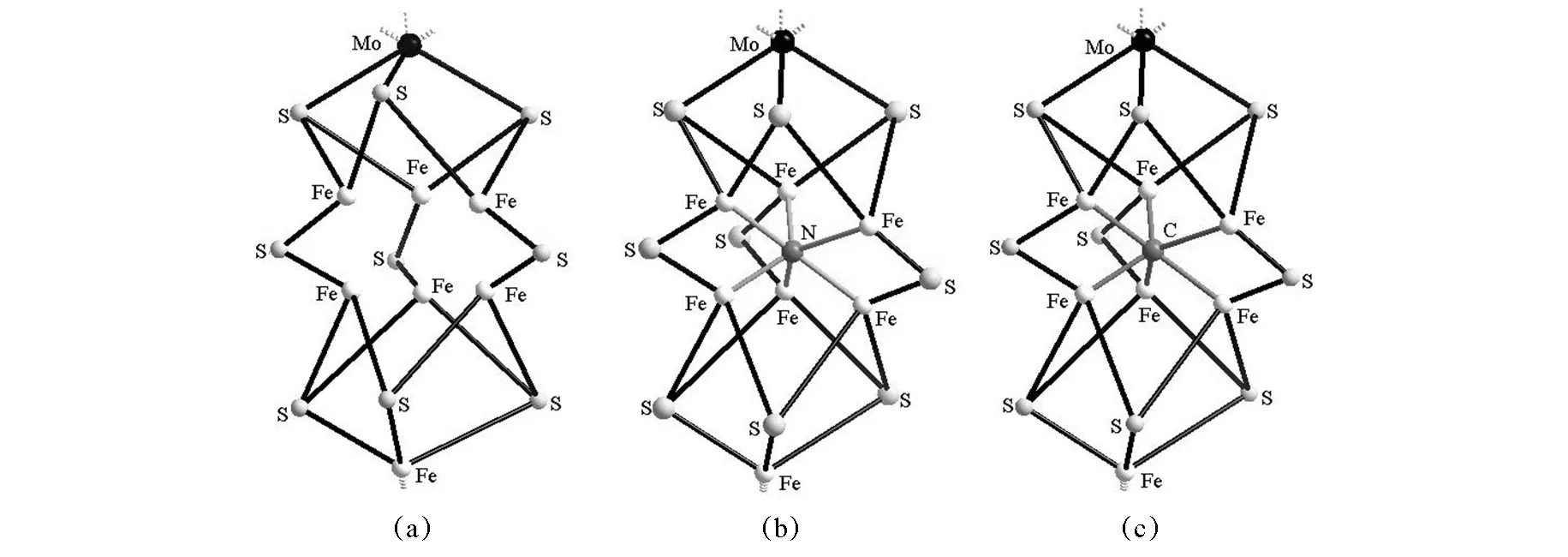
图7 固氮酶的结构研究
2.2.4 人工模拟固氮
人工模拟固氮的研究是在了解了生物固氮的上述内容后,在尽量接近生物条件下,人工模拟固氮酶的模型化合物合成并应用到工业生产中的研究。显然,研究集中在FeMo辅基的模拟合成上。虽然这种研究早在20世纪70年代就已开始,但只有当FeMo辅基的结构被确定之后,才给人工模拟合成指明了方向。
由于在FeMo辅基中有多个不饱和配位的Fe以及Mo原子处于FeMo辅基的外缘,这两点是FeMo辅基发挥固氮作用的关键,也是人工模拟合成中摆在化学家面前的难题。大体研究如下所述:
① 张纯喜[16]认为有两种途径有望克服这一困难。一种是引入体积较大的配体,合成低配位的Fe。在这方面Ell ison等[46]已作了尝试并得到了二配位和三配位的Fe的化合物。但这一方法有一定的局限性,因为体积大的配体所造成的体积效应阻碍了不饱和配位的Fe与N2等小分子的进一步作用。另一种是引入“保护配体”,利用它暂时稳定Fe的配位环境,在催化条件下,“保护配体”离去,从而产生不饱和配位的Fe。在许多无机和有机配体中,PR3可能是一类好的“保护配体”。目前人们对PR3类配体在FeMo辅基的模拟合成中的应用普遍较为重视[47-49]。
② 在FeMo辅基中,Mo原子处于FeMo辅基的外缘,这种不对称的结构对于合成有较大困难。从固氮酶自身的进化[50]可知,在固氮酶的进化过程中,首先是全Fe固氮酶,再逐步进化到含V固氮酶和含Mo固氮酶。可以先合成固氮酶的低级形式全Fe固氮酶,将另一端的Fe用其他金属取代,如用Mo、V等取代FeMo辅基或FeV辅基中另一端的Fe,这样就能简化FeMo辅基的人工模拟合成。
③ 由FeMo辅基活化还原底物N2的作用方式(图6)可以看出,Fe6S9[51-54]和Mo2Fe4S9[55]这两类化合物在骨架结构上(图8)与FeMo辅基最为相似,这正是FeMo辅基的重要特征。这可能是实现FeMo辅基人工模拟合成的有效途径。
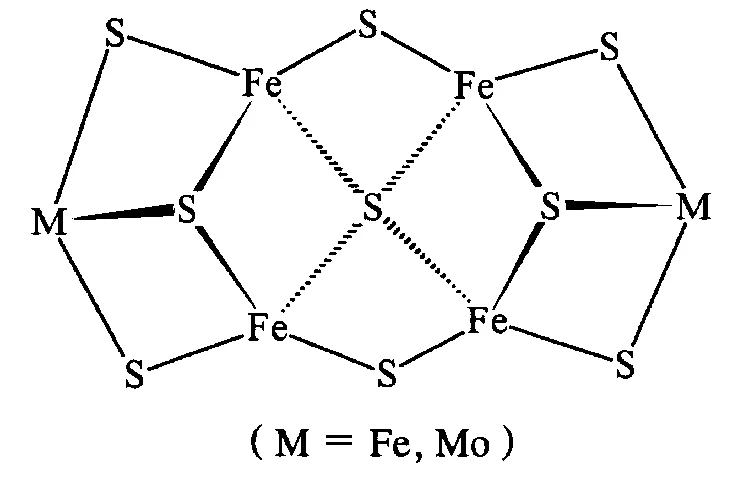
图8 模拟化合物骨架结构
这些也是当前化学模拟生物固氮的研究策略,最好是采用类似图8结构的簇合物作为催化剂,适当提高反应的温度(300 ℃以下)和压力(5.66 kPa以下),以利于H2的活化、削弱N2的三重键以及正反应的进行;而且不必改变现用设备,即可达到节约能源、降低生产成本、增加经济效益的目的[19]。
分子氮配合物又称双氮配合物(dinitrogen complex),是指含有分子氮作为配体的配合物。已制得的数以百计的双氮配合物几乎涵盖了元素周期表从ⅣB到Ⅷ族所有的过渡元素。其中能获得稳定分子氮配合物的元素有:Ti、Mo、W、Mn、Re、Fe、Co、Ni、Ru、Rh、Os、Ir、Pt,这些元素多为后过渡元素,它们在分子氮配合物中都处于低氧化态,因而含d电子较多。后来,由稀土元素[56]、金属卟啉[57]等物种也得到了其分子氮配合物。人们希望通过形成双氮配合物来削弱氮分子的三键、活化分子氮,从而达到将氮还原为氨的目的。较详细的介绍可参看一些综述文章[58-63]。
2.3.1 分子氮配合物的合成
分子氮配合物合成的背景可追溯到Vol’pin 和Shur的研究[64-66]:在过渡金属(Ti、Zr、V、Nb、Cr、Mo、W、Mn、Fe、Co)存在下,N2与还原剂如金属(Li、Mg、Al)、金属氢化物(LiAlH4)或金属有机化合物反应,可被还原成氨衍生物(通常为氮化物)。1965年,Allen和Senoff试图在水溶液中用水合肼还原三氯化钌制备[Ru(NH3)6]2+时,意外得到了第一个分子氮配合物[Ru(N2)(NH3)5]Cl2[67-68]:
该配合物的红外光谱在2100 cm-1处有一强吸收带。这一发现震惊了化学界,打破了长期以来认为氮分子不能作为电子对接受体,因而不能形成配合物的传统观念。从此,新的分子氮配合物不断涌现,以致成为热门研究课题。
分子氮配合物合成的途径主要有以下3种[58,61-63]:
① 直接法:用氮气和金属配合物在强还原剂存在下直接进行反应。例如:
如果金属配合物的配体为活性配体,则可在温和条件下直接被氮分子取代,并且反应可逆:
② 间接法:将含N—N键的配体转变成N2,这是大多数分子氮配合物的合成途径。例如:
③ 取代法:通过取代反应将一种分子氮配合物变成另一种分子氮配合物。例如:
此外还有其他合成方法。例如,由低温光解羰基化合物可产生不稳定的分子氮配合物:在-79 ℃下,用紫外光光解溶于液态Xe-N2混合物的Cr(CO)6,可产生一系列的Cr(CO)5-x(N2)配合物,产物已被IR光谱证实[69]。
2.3.2 分子氮配合物的配位方式和化学键
在分子氮配合物中,氮分子通常以端基或桥式端基的形式与金属原子结合,少数以侧基或桥式侧基的形式出现(图9)。
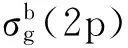
图9 氮分子和金属原子的几种配位方式
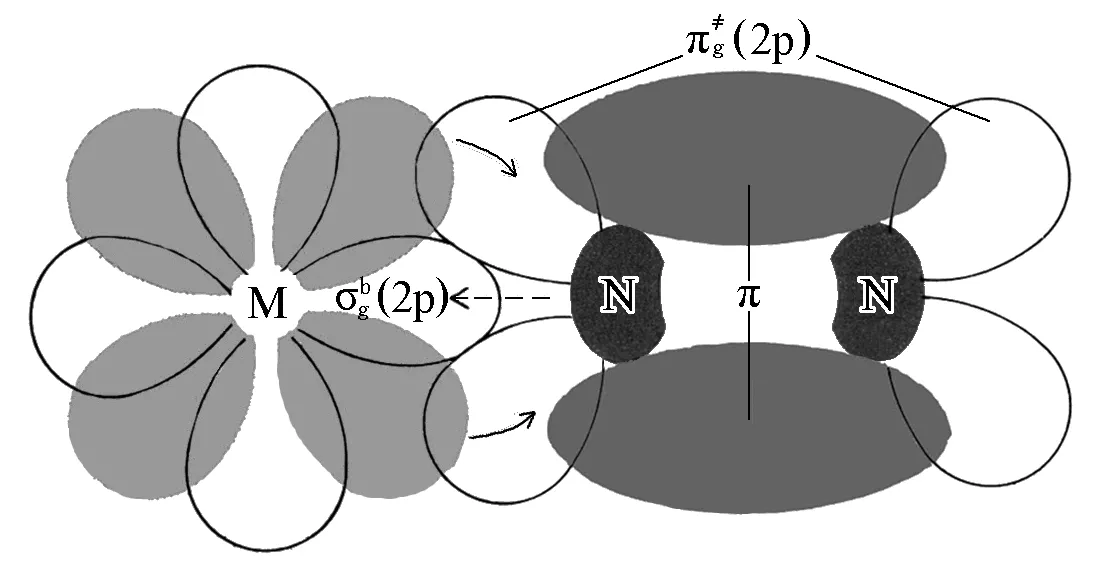
图10 M—N2键示意图
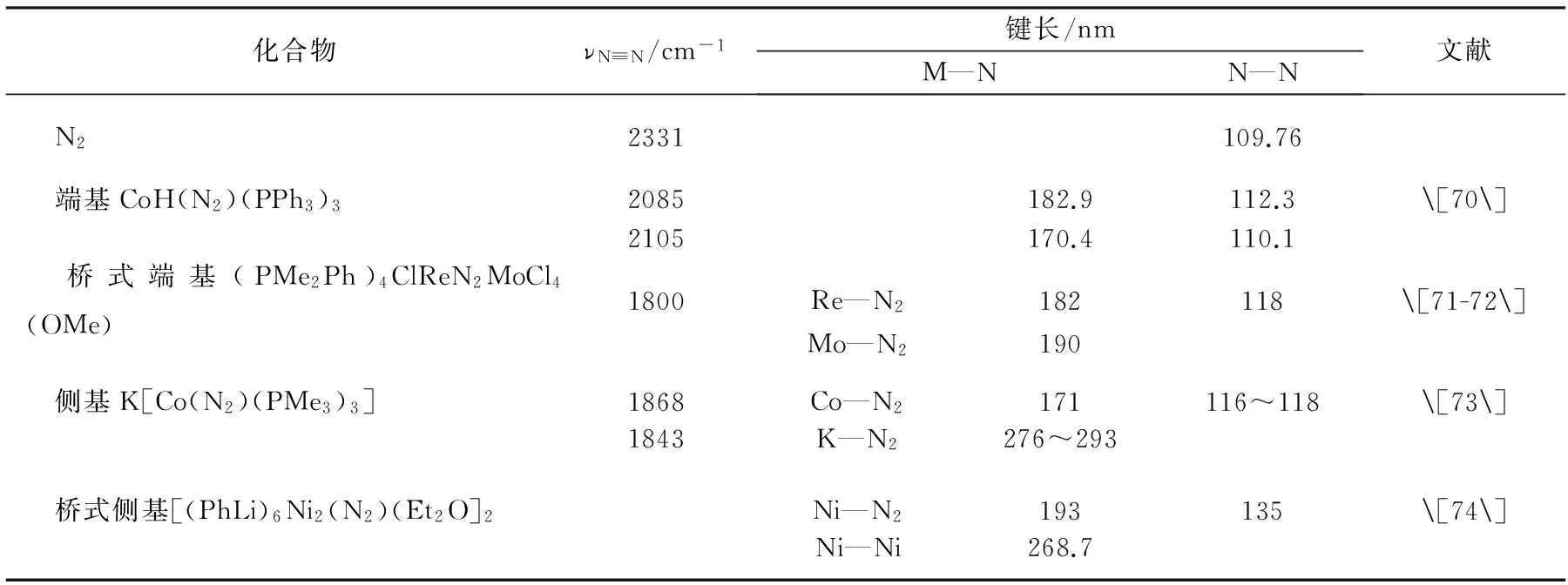
的部分结构数据
2.3.3 利用分子氮配合物的还原反应固氮

① 1975年,Chatt在20 ℃无氧条件下,在甲醇溶液中用硫酸处理顺-[M(N2)2(PMe2Ph)4],得到氨(表2)[75-77]:
收率可达90%。若用其他酸也可产生氨及联氨,但不如硫酸有效。
② 1988年,Taqui Khan等利用可见光激发,于水溶液中,以半导体Cd/Pt/RuO2为催化剂,在30 ℃和1×105Pa N2条件下,使[Ru(Hedta)N2](Hedta = trianion of ethylenediamine-tetraacetic acid)中的配位氮转化为NH3[78]。
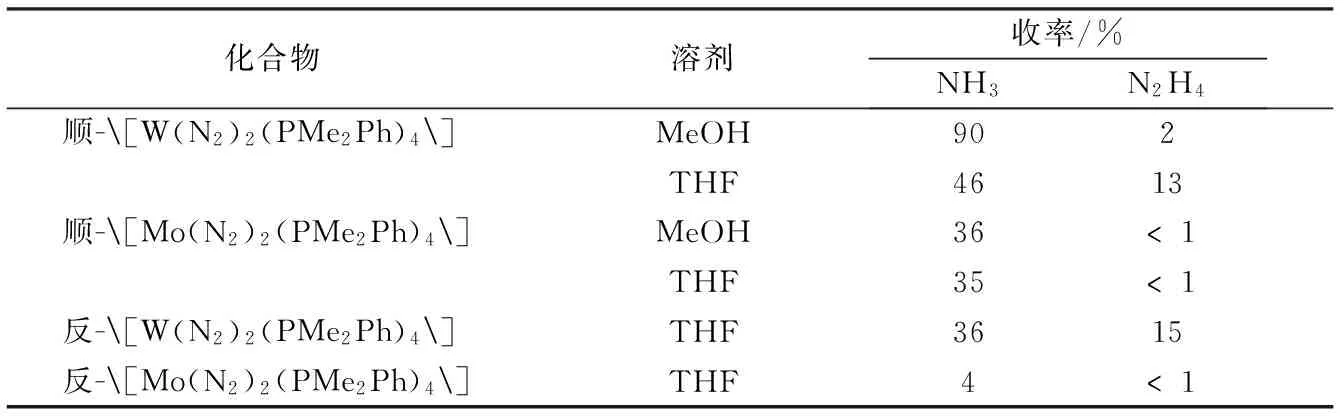
表2 钼、钨分子氮配合物与硫酸反应产生的氨及联氨的收率
③ 1992年,忻飞波等将在氦气氛中获得的分子氮配合物K3[Co(CN)5N3](按文献[79]制备)置于氢气氛中,研究其热分解情况,结果发现中间产物K3[Co(CN)5N2]和K6[Co(CN)10N2]在220 ℃得到NH3和HCN而不是N2[80]。这一结果为分子氮配合物的加氢成氨提供了有用信息。
值得一提的是:1995年,Laptaza等在常压和低于室温的条件下使N2与Mo(Ⅲ)配位,在甲苯或乙醚溶液中得到N3-离子配位的配合物:
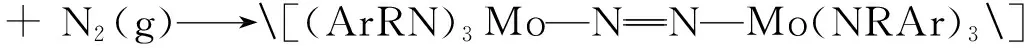
式中R为C(CD3)2CH3,Ar为3,5-C6H3(CH3)2;方括号中是假定的反应中间体。这项研究展示了一个奇迹:在温和条件下完成了N2分子中三重键的断裂。他们的成功显然与Mo(NRAr)3这个3配位化合物中配位不饱和Mo原子显示的路易斯酸性和Mo(Ⅲ)的给电子能力有关。
④ 1998年,Yoshiaki Nishibayashi等[81]的研究进展更加引人注目。在常压和55 ℃的条件下,通过分子氮配合物与分子氢配合物的反应产生了氨,收率达到55%(图11,表3)。这项研究在思路上的特点是,同时活化作为起始物的N2分子和H2分子:
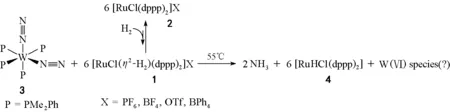
图11 分子氮配合物与分子氢配合物的反应生成氨的反应路线图
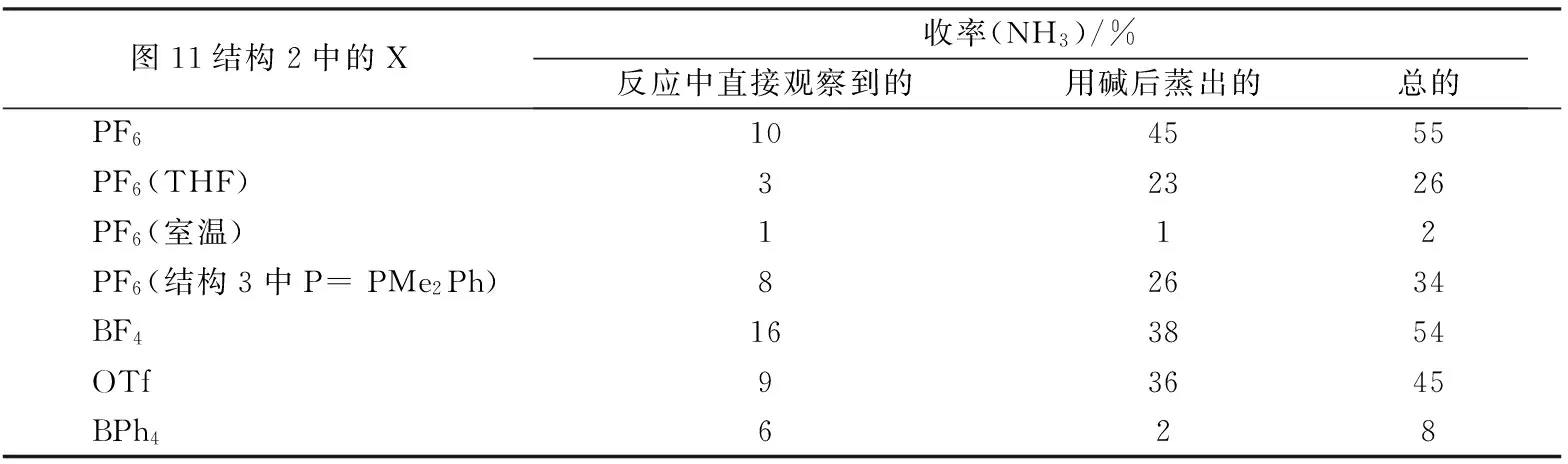
表3 不同X时的NH3的收率
⑤ 2010年,D.J.Knobloch等利用合成的铪氮分子配合物在室温下与CO反应,得到了最普通的化肥——草酰铵[82],收率为90%~95%。这项研究的最大特点是,同时使用了两种丰富原料作为反应物,在常温下将两个同样具有很强三键的N2和CO分子用最廉价的方法裂解得到普通的化肥。
除了上面两种可以弱化N≡N的思路以外,形成聚合氮结构也是可行的方法之一。在高压下,双原子氮会显示出丰富的化学键断裂和重组的现象,从而极大地改变其性质。理论计算和实验都已证明:压力能够改变氮原子与氮原子之间的距离。当分子间的距离与分子内的距离变得可以相比拟的时候,就会由双原子分子转化成由单键或键合的层状、链状、网状结构聚合氮的形式[83-91]。例如,实验室已制得N3和N4[92];被预测出的六氮苯(N6,类似于苯)被预言为高度不稳定(可能源自氮原子上相互排斥或能供电子给σ反键轨道的孤电子对)[93]和八氮立方烷(N8,类似于立方烷)被推测因为轨道对称的原因会动力学稳定[94]。有研究表明它们将是一种能量密度非常高的能源材料,如八氮立方烷预计拥有约22.9 MJ/kg的能量密度(假设完全分解为N2),是TNT的5倍[95]。从而引起科学家的广泛兴趣。
3 结束语
本文并非试图完成一篇有关活化固氮的综述,而是想在元素化学的教学中,探索一种教学方法。本意在于以讲解氮化学为例,从学生最为熟悉的氨的合成深入认识氮分子化学键的稳定性和活化固氮。 ① 在无机化学元素部分的教学中,我们强调要注重化学原理对元素化学的指导和应用作用[96]。N2是个有特点的无机分子,涉及的化学原理丰富多彩,包括化学键的价键理论、分子轨道理论分析,化学键稳定性的两面性等,对其较为全面的分析对如何学习其他元素具有示范效应。 ② 我们一直提倡在基础课教学中要重视对学生科研意识的培养。通过N2的分子配合物及其活化固氮研究进展的简介,都能起到引导学生灵活学习和运用知识去认识自然世界,提高分析问题和解决问题的能力的作用。 ③ 我们还注意引导一年级学生关注学术报告。初步学习如何查阅文献是我们培养学生热爱化学、增加学习热情和提高科学研究素养的一项基本教学举措。
教学探索实践证明[97],将教学与科学研究紧密结合的教学方法,有助于充分发挥教学方法本身的教育功能。
[1] Theodore G.The Elements:A Visual Exploration of Every Known Atom in the Universe.New York:BlackDog & Leventhal,2009
[2] Haynes W H.Handbook of Chemistry and Physics.91st ed.Boca Raton:CRC,2010-2011
[3] Al-Joboury M I,May D P,Turner D W.JChemSoc,1965,44:44616
[4] Turner D W,May D P.JChemPhys,1966,45:471
[5] Mulliken R S.CanJChem,1958,36:10
[6] Sellmann D.AngewChemIntEd,1974,13(10):639
[7] Heidemann.ChemZtg,1921,45:1073
[8] Andersen E B.ZPhysik,1922,10:54
[9] Storch H H,Olson A R.JAmChemSoc,1923,45:1605
[10] Modak J M.Resonance,2002,7:69
[11] Freund H J,Knozinger H.JPhysChemB,2004,108(38):14183
[12] Ertl G,Vac J.SciTechA,1983,1:1247
[13] 马秀芳,邓辉球,李微雪.科技导报,2007,25(24):25
[14] Kitano M,Inoue Y,Yamazaki Y,etal.NatureChem,2012,4:934
[15] 徐晔,张金池,王广林,等.生物学杂志,2011,28(4):61
[16] 张纯喜.化学进展,1997,9(2):131
[17] 黄静伟,张鸿图,万惠霖,等.生物化学与生物物理进展,1996,23(1):18
[18] 周朝晖,颜文斌,张凤章,等.厦门大学学报(自然科学版),2001,40(2):321
[19] 王友绍,李季伦.自然科学进展,2000,10(6):482
[20] Burris R H.JBiolChem,1991,266(15):9339
[21] Robson R L,Eady R R,Richardson T H,etal.Nature,1986,322:388
[22] Simpson F B,Burris R H.Science,1984,224:1095
[23] Johnson W H O.Science,1992,257:1639
[24] Kim J,Rees D C.BiochemJ,1994,33(2):389
[25] Thoreley R N F,Lowe D J.BiochemJ,1983,215(2):393
[26] Thoreley R N F,Lowe D J.Kinetics and Mechanism of the Nitrogenaseenzyme System.New York:Weily,1985
[27] Chan M K,Kim J,Rees D C.Science,1993,260:792
[28] Deng H,Hoffmann R.AngewChemIntEd,1993,32(7):1062
[29] Dance I G.AustJChem,1994,47(5):979
[30] Zhong S J,Liu C W.Polyhedron,1997,16(4):653
[31] 吴新涛,卢嘉锡.科学通报,1995,40(7):577
[32] Tsai K R,Wan H L.JClusterScience,1995,6(4):485
[33] 黄静伟,张风章,许良树,等.高等学校化学学报,1995,16(6):920
[34] Shah V K,Brill W J.ProcNatlAcadSci, 1977,74(8):3249
[35] Camahan J E,Morteson L E,Mower H F,etal.BiochimBiophysActa,1960,38:188
[36] Camahan J E,Morteson L E,Mower H F,etal.BiochimBiophysActa,1960,44:520
[37] Morteson L E.Nitrogen Fixatin in Extracts of Clostridium Pasteuranium.Ohio:Antioch Press,1965
[38] Kennedy C K,Eady P R,Kondorpsi E,etal.BiochemJ,1976,155:383
[39] Kim J,Rees D C.Nature,1992,360:553
[40] Kim J,Rees D C.Science,1992,257:1677
[41] Kim J,Woo D,Rees D C.Biochem,1993,32:7104
[42] Kim J,Rees D C.Biochem,1994,33:389
[43] Einsle O,Tezcan F A,Andrade S L A,etal.Science,2002,297:1696
[44] Kyle M,Lancaster M R,Patrick E,etal.Science,2011,334:974
[45] Thomas S,Müge A,Limei Z,etal.Science,2011,940
[46] Ellison J J,Ruhlandt-Senge K,Power P P.AngewChemIntEd,1994,33(11):1178
[47] Scott M J,Holm R H.AngewChemIntEd,1993,32(4):564
[48] Demadis K D,Campana C F,Coucouvanis D J.JAmChemSoc,1995,117(29):7832
[49] Cen W,MacDonnell F M,Scott M I,etal.InorgChem,1994,33(25):5809
[50] Leigh G J,Jimenez-Tenorio M J.JAmChemSoc,1991,113(15):5862
[51] 张致贵,刘喜生,樊玉国.科学通报,1985,30(12):913
[52] Strasdeit H,Krebs B,Henkel G.InorgChem,1984,23(13):1816
[53] Hagen K S,Watson A D,Hlm R H.JAmChemSoc,1983,105(12):3905
[54] Christou G,Sabat M,Ibers J A,etal.InorgChem,1982,21(9):3518
[55] 张致贵,樊玉国,李宇,等.科学通报,1986,31(21):1634
[56] Evans W J,Ulibarri T A,Ziller J W.JAmChemSoc,1988,110:6877
[57] Camenznd M J.ChemCommun,1986,1137
[58] Sellmann D.AngewChemIntEd,1974,13:639
[59] Chatt J,Leigh G J.ChemSocRev,1972,1:121
[60] Dallen A,Harria R O,Loescher B R,etal.ChemRev,1973,11:73
[61] Chatt J,Dilworth J R,Richards R L.ChemRev,1978,78:589
[62] 项斯芬,严宣申,曹庭礼,等.无机化学丛书.第四卷.北京:科学出版社,1995
[63] 席振峰,金斗满.化学通报,1991(3):1
[64] Vol’pin M E,Shur V B.DokladyAkadNaukSSSR,1964,156:1102
[65] Vol’pin M E,Shur V B,Vestnik.DokladyAkadNaukSSSR,1965,157:51
[66] Vol’pin M E,Shur V B,Bichin L P.DokladyAkadNaukSSSR,1965,157:720
[67] Allen A D,Senoff C V.ChemCommu,1965:621
[68] 吴永仁.中国中学教学百科全书-化学卷.沈阳:沈阳出版社
[69] Turder J J,Simpion M B,Poliakoff M,etal.InorgChem,1986,22:911
[70] Davis B R,Payne N C,Ibert J A.InorgChem,1969,8:2719
[71] Merceer M,Crabtrec R H,Ricbards R L.ChemCommun,1973:808
[72] Merceer M.DoltonTran,1974:1637
[73] Hammer R,Klein H F,Friedrich P,etal.AngewChemIntEd,1977,16:485
[74] Kroger C,Tsay Y H.AngewChemIntEd,1973,12:998
[75] Chatt J,Pearman A J,Richards R L.DoltonTrans,1977,1853
[76] Chatt J,Pearman A J,Richards R L.Nature,1975,253:39
[77] Collman J,Hutchinson J E,Lopez M A,etal.JAmChemSoc,1991,113:2794
[78] Taqui Khan M M.AngewChemIntEd,1988,27:923
[79] Nast R,Ronner M.ZAnorgChem,1956,285:278
[80] 忻飞波,郑丽敏,忻新泉.无机化学学报,1992,8(1):82
[81] Nishibayashi Y,Iwai S,Hidai M.Science,1988,279:540
[82] Knobloch D J,Lobkovsky E,Chirik P J.NatureChem,2010,2:30
[83] Mailhiot C,Yang L H,McMahan A K.PhysRevB,1992,46:14419
[84] Zahariev F,Hu A,Hopper J,etal.PhysRevB,2005,72:214108
[85] Martin R M.PhysRevLett,2003,91:105501
[86] Eremets M I,Gavriliuk A G,Trojan I A,etal.NatMater,2004,3:558
[87] 王晓丽.高压下固态氮的相变研究与新型碳氮超硬材料的理论设计.长春:吉林大学,2011
[88] Wang X L,Bao K,Tian F B,etal.JChemPhys,2010,133:044512
[89] Wang X L,Bao K,Wang L C,etal.JChemPhys,2010,132:024502
[90] Wang X L,He Z,Ma Y M,etal.JPhysCondensMatter,2007,19:425226
[91] Wang L C,Bao K,Meng X.JChemPhys,2011,134:024517
[92] Polymeric Nitrogen Synthesized.http://phys.org/news693.html,2004-08-05/2014-06-16
[93] Fabian J,Lewars E.CanaJChem, 2004,82(1):50
[94] U N Patil,Dhumal N R,Gejji S P.TheoreChimActa,2004,112:27
[95] Glukhovtsev M N,Jiao H J,Schleyer P R,etal.InorgChem,1996,35:7124
[96] 谢钢,杨奇,陈三平,等.大学化学,2012,27(5):36
[97] 陈三平,谢钢,杨奇,等.大学化学,2012,27(3):13
Learning the Stability and Activation of Chemical Bond of the Nitrogen Molecule from Ammonia Synthesis*
Yang Qi Xie Gang Chen Sanping**Zhang Yifan Guo Peiyu Gao Shengli
(CollegeofChemistryandMaterialsScience,NorthwestUniversity,Xi′an710069,Shaanxi,China)
This paper discusses the stability of the nitrogen molecule based on the chemical bond, and then introduces researches related to the nitrogen chemical bond activation from the industrial ammonia synthesis. The authors desire to explore a new approach for teaching elemental chemistry. The aim is to lead students to study and use the knowledge to learn the natural world, and improve the ability to analyze and solve problems.
Ammonia synthesis; Nitrogenase; Nitrogen complex; Teaching method
10.3866/pku.DXHX20150633
*通讯联系人,E-mail:sanpingchen@126.com 张一凡 郭培宇 高胜利
第三批国家级精品资源共享课(教高函[2013]132号);国家级教学团队《无机化学和分析化学基础课教学团队》(教高函、[2007]23号);国家基础科学人才培养基金(No.J1210057)
O6; G64
