复方柴胡袋泡茶的质量标准*
2015-01-05许本善雷宁陈靖婕常子倩马萍童卫杭
许本善,雷宁,陈靖婕,常子倩,马萍,童卫杭
(解放军第二炮兵总医院药学部,北京 100088)
复方柴胡袋泡茶的质量标准*
许本善,雷宁,陈靖婕,常子倩,马萍,童卫杭
(解放军第二炮兵总医院药学部,北京 100088)
目的完善复方柴胡袋泡茶的质量控制标准,保障制剂质量。方法采用薄层色谱(TLC)法对制剂中的柴胡、葛根、连翘进行定性鉴别,用高效液相色谱(HPLC)法对制剂中的葛根素进行含量测定。色谱条件:色谱柱为岛津VP-ODS-C18柱(250 mm×4.6 mm,5 μm),流动相为甲醇-0.2%磷酸溶液(23:77),流速为1.0 mL·min-1,柱温为30 ℃,检测波长为250 nm。结果TLC斑点清晰,分离度良好,阴性对照无干扰;葛根素线性范围为1.340 8~56.632 8 μg·mL-1(r=0.999 9,n=6),平均加样回收率102.98%,RSD为1.48%(n=6)。结论该方法简便易行,适用于复方柴胡袋泡茶的质量控制。
柴胡袋泡茶,复方;葛根素;色谱法,薄层;色谱法,高效液相;含量测定
复方柴胡袋泡茶系我院医院制剂,具有解肌发表,清热解毒,清利头目,发散风热,清理除烦之功效,适用于流感、普通感冒及肺炎、化脓性扁桃体炎等引起的发热。该制剂处方由柴胡、葛根、黄芩、青蒿、连翘、浙贝、生石膏等12味药材组成,来源于我院多年临床经验,疗效确切。该制剂原质量标准制定于上世纪90年代,未对其中主要药味及其活性成分进行定性、定量检测。为了更好地控制制剂质量,本研究增加了制剂中柴胡、葛根、连翘的薄层鉴别,同时建立高效液相色谱法对制剂中葛根素的含量进行测定。实验结果表明,所建方法操作简便、灵敏度好、重复性佳,可用于该制剂的质量控制。
1 仪器与试药
1.1 仪器 超声波清洗器(KQ-500V型,昆山超声仪器有限公司);高效液相色谱系统(日本岛津LC-20A,包括DGU-20A3型脱气机、LC-20AT型溶剂输送泵、SIL-20A型自动进样器、CTO-20A型柱温箱、SPD-20A型紫外-可见检测器,以及LC Solution色谱工作站);电子天平(JM-B20002型,浙江省余姚市纪铭称重校验设备有限公司,感量:0.1 mg);分析天平(Sartorius,CP225D型,感量:0.01 mg);旋转蒸发器(RE-52AA型,上海亚荣生化仪器厂)。
1.2 试药 硅胶G薄层板(青岛海洋化工厂);柴胡皂苷a对照品(中国食品药品检定研究院,供含量测定用,批号:110777-201108,含量:90.50%)、柴胡皂苷d对照品(中国食品药品检定研究院,供含量测定用,批号:110778-201208,含量:94.60%)、葛根素对照品(中国食品药品检定研究院,供含量测定用,批号:110752-201313,含量:95.50%)、连翘苷对照品(中国食品药品检定研究院,供含量测定用,批号:110821-201213,含量:95.30%);复方柴胡袋泡茶[医院制剂,批准文号:总制字(2011)F07002,批号:20130417,20131216,20140418];甲醇为色谱纯,水为灭菌注射用水,其余试剂均为分析纯。
2 方法与结果
2.1 薄层色谱鉴别
2.1.1 柴胡 取制剂粉末(批号:20140418,过筛孔内径0.250 mm筛)6.5 g,置具塞锥形瓶中,加5%浓氨水试液的甲醇溶液100 mL,密塞,30 ℃水温超声处理30 min,滤过,用甲醇分2次洗涤容器及药渣,洗液与滤液合并,回收溶剂至干。残渣加20 mL水溶解,再用饱和正丁醇提取,反复提取3次,每次50 mL,合并正丁醇液,用水洗涤2次,每次20 mL,弃去水液,回收正丁醇至干,甲醇溶解,并定容至5 mL,摇匀,作为供试品溶液。另取北柴胡对照药材0.5 g,同法制成对照药材溶液。取柴胡皂苷a和柴胡皂苷d的对照品适量,分别加甲醇制成每毫升含0.2 mg的溶液,作为对照品溶液[1]。照薄层色谱法[2010年版《中华人民共和国药典》一部附录ⅥB]实验,吸取上述供试品溶液和对照品溶液各1 μL,分别点于同一硅胶G薄层板上,以乙酸乙酯-乙醇-水(8:2:1)为展开剂,展开,取出,晾干,喷以2%对二甲氨基苯甲醛的40%硫酸溶液,于60 ℃下加热至斑点显色清晰,紫外光灯(365 nm)下检视。供试品色谱中,在与对照品色谱相应的位置上,显相同的黄色荧光斑点,阴性样品无干扰(图1)。

1,2.样品;3.柴胡皂苷a;4.柴胡皂苷d;5.北柴胡对照药材;6,7.阴性样品
图1 柴胡的薄层色谱图(温度:4 ℃;相对湿度:50%)
1,2.sample;3.saikosaponin a;4.saikosaponin d;5.reference ofBupleurumchinenseDC.;6,7.negative sample
Fig.1 TLC chromatogram ofBupleurumchinenseDC. (temperature: 4 ℃; relative humidity: 50%)
2.1.2 葛根 取制剂粉末(批号:20140418, 过筛孔内径0.300 mm筛)10 g,加甲醇100 mL,放置2 h,滤过,滤液蒸干,残渣加甲醇5 mL使溶解,作为供试品溶液。另取葛根对照药材0.8 g,同法制成对照药材溶液。再取葛根素对照品,加甲醇制成每毫升含1 mg的溶液,作为对照品溶液[2]。照薄层色谱法[2010年版《中华人民共和国药典》一部附录ⅥB]实验,吸取上述溶液各2 μL,分别点于同一硅胶G薄层板上,使成条状,以二氯甲烷-乙酸乙酯-甲醇-水(15:40:22:5)为展开剂,展开,取出,晾干,置紫外光灯(254 nm)下检视。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光条斑。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的荧光斑点(图2)。
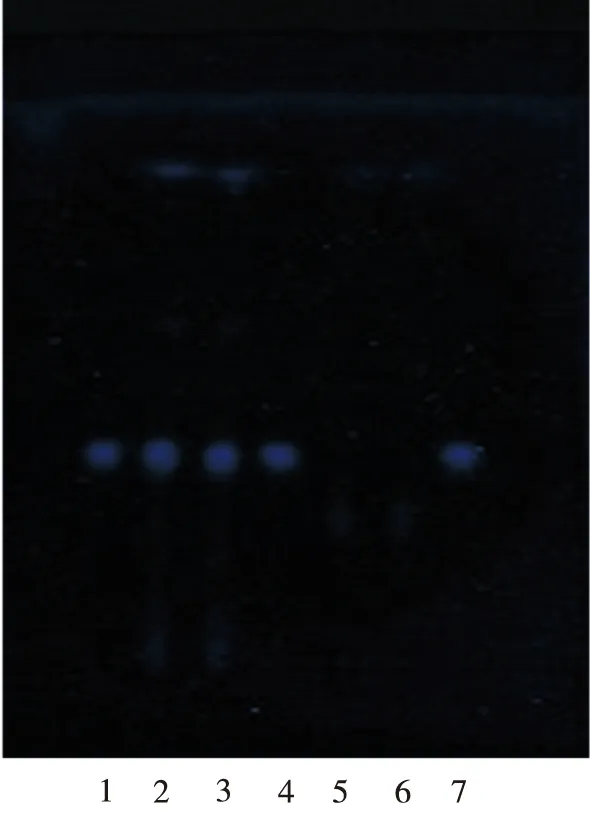
1,4.葛根对照药材;2,3.样品;5,6.阴性样品;7.葛根素对照品
图2 葛根的薄层色谱图(温度:4 ℃;相对湿度:50%)
1,4.RadixPuerariaeLobatae;2,3.sample;5,6.negative samples;7.puerarin reference
Fig.2 TLC chromatogram ofRadixPuerariaeLobatae(temperature: 4 ℃; relative humidity: 50%)
2.1.3 连翘 取制剂粉末(批号:20140418,过筛孔内径0.180 mm筛)15 g,加石油醚(30~60 ℃)50 mL,密塞,超声处理15 min,滤过,弃去石油醚液,残渣于通风橱中挥干石油醚,再加入甲醇50 mL,密塞,超声处理20 min,滤过,滤液蒸干,残渣加甲醇5 mL使溶解,作为供试品溶液[2]。再取连翘苷对照品,加甲醇制成每毫升含0.25 mg的溶液,作为对照品溶液。照薄层色谱法[2010年版《中华人民共和国药典》一部附录ⅥB]实验,吸取上述溶液各2 μL,分别点于同一硅胶G薄层板上,以乙酸乙酯-丙酮-甲酸-水(5:3:1:1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105 ℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。经阴性对照实验,结果阴性对照溶液无干扰(图3)。
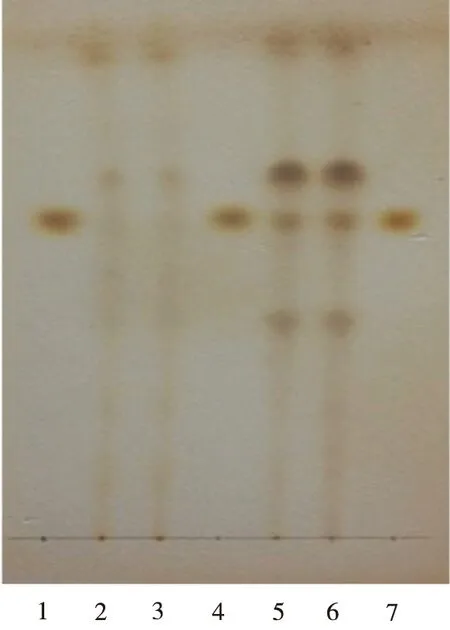
1,4,7.连翘苷对照品;2,3.阴性样品;5,6.样品
图3 连翘的薄层色谱图(温度:4 ℃;相对湿度:50%)
1,4,7.Phillyrinreference; 2,3. negative sample; 5,6. sample
Fig.3 TLC chromatogram ofForsythiasuspensa(temperature: 4 ℃; relative humidity: 50%)
2.2 葛根素含量测定
2.2.1 色谱条件与系统适用性实验 色谱柱:岛津 VP-ODS-C18(250 mm×4.6 mm,5 μm);流动相:甲醇-0.2%磷酸溶液(23:77);检测波长:250 nm;流速:1.0 mL·min-1;柱温:30 ℃;进样量:10 μL。理论板数按葛根素峰计算应不低于4 500,分离度>1.5[3]。分别吸取对照品溶液、供试品溶液、阴性样品溶液,注入高效液相色谱仪,测定,供试品与对照品在相同位置有吸收,阴性样品无干扰。
2.2.2 对照品溶液的制备 精密称取葛根素对照品6.25,12.50 mg分别置25 mL量瓶中,加甲醇稀释至刻度,摇匀作为储备液;精密量取0.25 mg·mL-1储备液1 mL置25 mL量瓶中,加甲醇稀释至刻度,摇匀,作为对照品溶液,浓度为10 μg· mL-1。
2.2.3 供试品溶液的制备 取制剂(批号:20130417)10袋,混匀,过筛孔内径0.300 mm筛,称取筛下制剂粉末约0.25 g,精密称定,置具塞锥形瓶中,精密加入30%乙醇50 mL,称定质量,超声(500 W,40 kHz)30 min,放冷,再称定质量,用30%乙醇补足减失的质量,摇匀,滤过,取续滤液,作为供试品溶液。
2.2.4 阴性样品溶液的制备 按处方量比例及制备工艺,取缺葛根的其他药材制备阴性样品。精密称取阴性样品,按照供试品溶液的制备方法制成阴性样品溶液。
2.2.5 线性关系考察 分别精密吸取葛根素对照品贮备液(0.25 mg· mL-1)0.5,1.0,2.0 mL至10 mL 量瓶中,用色谱纯甲醇定容,分别标记为4~6号。另分别取0.5 mL葛根素对照品贮备液(0.25 mg·mL-1)至100,50,25 mL量瓶中,甲醇定容,标记为1~3号。分别精密吸取上述溶液各10 μL,按“2.2.1”项下的色谱条件测定。以峰面积积分值(Y)对葛根素进样浓度(X)进行线性回归,得到葛根素的回归方程为:Y=43 347X+7 444.3(r=0.999 9,n=6)。结果表明,葛根素在1.340 8~53.632 8 μg·mL-1的浓度范围内,峰面积与浓度呈良好的线性关系。
2.2.6 精密度实验 取对照品溶液,重复进样6次,进样量10 μL,在上述色谱条件下求得葛根素峰面积的RSD为0.38%(n=6)。
2.2.7 重复性实验 取供试品(批号:20130417)共6份,分别按“2.2.3”方法制备供试品溶液,进行测定,求得葛根素含量的RSD为1.10%(n=6),表明重复性较好。

A.阴性对照溶液;B.对照品溶液;C.样品溶液;1.葛根素
2.2.8 稳定性实验 取同一供试品溶液,在0,1,2,3,5,10 h分别进行测定。结果表明样品溶液在10 h内基本稳定,葛根素峰面积的RSD为0.16%(n=6)。
2.2.9 加样回收率实验 精密称取已知含量(批号:20130417)的样品约0.12 g,共6份,分别置具塞锥形瓶中,分别精密加入葛根素对照品贮备液(0.50 mg·mL-1)1 mL,按“2.2.3”项下方法制备即得。精密吸取上述供试液10 μL注入色谱仪,测定含量,结果平均回收率102.98%(RSD=1.48%,n=6)。见表1。
表1 葛根素加样回收率实验结果
Tab.1 Results of recovery test of puerarin

取样量/g原有量加入量测得量mg回收率/%0.12120.41450.48710.9140102.550.12140.41520.48710.9072101.010.12140.41520.48710.9243104.520.12140.41520.48710.9270105.070.12170.41620.48710.9141102.220.12130.41480.48710.9141102.50
2.2.10 样品含量测定 取3批(批号:20130417,20131216,20140418)复方柴胡袋泡茶制剂,按“2.2.2”及“2.2.3”项下方法分别制备对照品溶液和供试品溶液,按上述色谱条件进行测定并计算含量。结果葛根素含量依次为3.42,3.28,3.35 mgg-1,RSD分别为2.43%,2.69%,1.10%。
3 讨论
3.1 薄层鉴别药材的选择 在薄层鉴别过程中还对处方中除了柴胡、葛根、连翘以外的其他9味药材进行了考察,均无法排除阴性样品的干扰,故未纳入质量标准。
3.2 含量测定指标性成分的选择 本研究最初选用黄芩苷作为制剂含量测定的指标性成分,先后选用“加
热回流”[2]、“超声提取”[4]、“AB-8大孔树脂分离”[5]等前处理方法摸索供试品的制备,同时优化色谱条件,但发现其阴性样品中始终存在不同程度的干扰,故重新选取葛根素为含量测定的指标性成分。
3.3 流动相的选择 实验中曾考察了以甲醇-水(25:75)为流动相,但葛根素分离度和峰形均不佳[2];而以甲醇-0.2%磷酸溶液(23:77)为流动相时,葛根素峰形对称,分离度良好[6]。
3.4 含量测定中供试品制备条件的选择 首先考察了回流、超声两种提取方法,结果显示超声提取的效果较好且操作简便;其次,对提取溶剂(30%甲醇、30%乙醇)进行了考察,结果显示,30%乙醇提取效果稍优于30%甲醇,兼顾环保无毒,选择30%乙醇作为提取溶剂;第三,考察了超声提取时间,结果表明30 min以后样品中葛根素的含量不再增加。因此,最终确定供试品处理方法为30%乙醇超声处理30 min。
[1] 李竞,高英,王振华,等.柴胡的质量评价研究[J].北方药学,2013,10(6):8-10.
[2] 国家药典委员会.中华人民共和国药典(一部)[M].北京:中国医药科技出版社,2010:312-313,159-160,282-283.
[3] 林清,吴雪梅,马晓鹏.高效液相色谱法测定风寒感冒颗粒中葛根素的含量[J].医药导报,2009,28(9):1211-1212.
[4] 李景松,张贵军,张智圆,等.黄芩药材中活性成分黄芩苷的研究概况[J].世界中医药,2013,8(4):469-471.
[5] 刘希博,段姚尧,潘欣,等.大孔树脂精制黄芩中黄芩苷的工艺研究[J].武警后勤学院学报(医学版),2013,22(6):473-475.
[6] 熊汝菊,马新飞,陆兔林,等.高效液相色谱法测定颈宁胶囊中葛根素的含量[J].中国中医药信息杂志,2008,15(2):53-54.
《临床外科杂志》征订启事
《临床外科杂志》是由中华医学会湖北分会主办,全国公开发行的临床外科专业性学术类期刊,为国家科学技术部中国科技论文统计源期刊(中国科技核心期刊)。并荣获首届《CAJ-CD规范》执行优秀期刊奖。国际标准刊号:ISSN1005-6483,国内统一刊号:CN42-1334/R。本刊以“面向临床,指导临床,服务临床”为宗旨,报道外科领域中最新的科研成果和临床诊治经验,密切结合临床实践,对外科临床和科研工作有很强的指导作用。辟有述评、专家笔谈、论著、术式介绍、技术革新、讲座、综述、会议(座谈)纪要、临床病例讨论、教学查房、学术争鸣、国内外学术动态、基层医院经验、病例报告、问题解答、新期刊文献、书评等栏目。
《临床外科杂志》为月刊。欢迎广大读者及时到当地邮局订阅(邮发代号38-184),2016年每册定价10.00元(每期80页)。如错过邮局订阅时间,可随时向本刊编辑部邮购。编辑部地址:湖北省武汉市武昌区东湖路165号,邮政编码:430071,联系电话:027-87893476,传真:027-87893476,Email:whlcwk@126.com,http://www.lcwkzz.com。
Quality Standard for Compound Chaihu Tea
XU Benshan, LEI Ning, CHEN Jingjie, CHANG Ziqian, MA Ping, TONG Weihang
(DepartmentofPharmacy,theSecondArtilleryGeneralHospitalofChinesePeople'sLiberationArmy,Beijing100088,China)
Objective To improve the quality standard of compoundChaihutea. MethodsPupleuriRadix,PuerariaeLobataeRadixandForsythiaeFructuswere identified by thin layer chromatography (TLC).HPLC method was used to determine the content of puerarin.The determination was performed on aShimadzuVP-ODS-C18column (250 mm×4.6 mm,5 μm) with the mobile phase of methanol-0.2% H3PO4(23:77).The flow rate was 1.0 mL·min-1, the column temperature was set at 30 ℃ and the detection wavelength was 250 nm. Results The TLC spots were clear and well-separated without negative interference.The good linear correlation existed within the range of 1.340 8-56.632 8 μg· mL-1for puerarin (r=0.999 9,n=6) and the average recovery was 102.98% with RSD being 1.48% (n=6). Conclusion The method is simple and accurate.It can be used for quality control of compoundChaihutea.
Chaihutea, compound; Puerarin; Chromatography, thin layer; Chromatography, high performance liquid; Content determination
2015-01-20
2015-03-04
*全军医疗机构制剂标准提高科研专项课题面上项目(13ZJZ19-2)
许本善(1987-),男,山东德州人,药师,学士,研究方向:中药质量控制。电话:(0)18911632870, E-mail:benshan.xu@163.com。
雷宁(1978-),女,陕西咸阳人,主管药师,博士,研究方向:天然药物药效物质基础及质量控制。电话:010-66343245(转8002),E-mail:lilyzebra@163.com。
R286;R927.2
B
1004-0781(2015)12-1640-04
10.3870/j.issn.1004-0781.2015.12.023
