同型半胱氨酸体内代谢及调节的研究进展
2015-01-04吕海宏汤旭磊
吕海宏 汤旭磊
目前研究认为同型半胱氨酸不仅是心脑血管疾病的独立危险因素,并且也认为是骨质疏松性骨折的独立危险因素。临床应用上,同型半胱氨酸主要作为心血管疾病,尤其是冠状动脉粥硬化和心肌梗塞的危险指标,它的浓度升高程度与疾病的危险性成正比;进一步研究认为同型半胱氨酸与脑血管疾病、高血压、骨质疏松和肾脏疾病也有密切相关性,因此同型半胱氨酸与临床疾病的关系日益引起人们的重视。
1 同型半胱氨酸(Hcy)代谢
人体内Hcy的来源是必需氨基酸-蛋氨酸(Met)的脱甲基作用,此过程产生2个中间化合物S-腺苷甲硫氨酸(AdoMet)和S-腺苷同型半胱氨酸(AdoHcy)[1]。因此,Hcy是一种含硫氨基酸,是蛋氨酸代谢的中间产物,是体内一碳单位代谢的一个正常中间产物。AdoMet 由MAT(ATP-L-methionine S-adenosyltransferase)催化生成,ATP的腺苷转移到MET,在核糖的5-C和氨基酸的S原子间产生1个硫酰基,形成一个高能复合物。MAT有2个不同的同工酶,其中一个是肝脏特异性的,另一个是组织非特异性的[2]。第1种与Met有相对高的Km,第2种与Met的Km 较低。研究表明,体内甲基团的利用主要存在肌酸形成反应中,它消耗的AdoMet比所有其它的转甲基反应的总和更多[3]。Hcy主要参与甲基转移的代谢,Hcy体内代谢包括4条途径:(1)甲基化途径(见图1)。在甲基化途径中,Hcy接受甲基四氢叶酸提供的甲基转化为甲硫氨酸,后者经活化生成AdoHcy,而AdoMet是一个活泼的甲基供体,是体内超过115种转甲基反应的重要甲基供体[4],包括DNA、RNA及脂质,常见的DNA甲基化 (DNA methylation)即以AdoMet为甲基供体,由甲基转移酶催化形成,是一种重要的表观修饰,在基因表达和调控中担当关键角色[5]。AdoMet捐出甲基后转变为AdoHcy,大多数AdoMet依赖的甲基转移酶可以被AdoHcy所抑制。AdoHcy在AdoHcy水解酶作用下水解而成腺苷酸和Hcy。AdoHcy水解酶广泛分布于哺乳动物各种组织中,是一种有效的甲基转移酶抑制剂,可抑制甲基化过程。AdoHcy水解酶为一可逆性反应酶,催化的反应是可逆的,但按照热力学平衡方程,平衡向合成AdoHcy的方向移动,而不是向水解的方向移动。不管哪种选择,AdoHcy都可以被细胞内结合蛋白移除,或者可能部分被移出细胞外。在体内,不管是Hcy还是腺苷常常被快速移除,以便水解酶向分解的方向作用。AdoMet/AdoHcy的比率常被作为细胞内甲基化的预测因子。体内四氢叶酸在5-二甲基四氢叶酸还原酶(5-MTHFR)的作用下生成N,CH 3-四氢叶酸,进一步提供1个甲基,以维生素(VitB12)为辅基,与Hcy反应再甲基化生成蛋氨酸,即为甲硫氨酸循环。(2)转硫化途径。此条途径是甲硫氨酸(Met)代谢的主要途径,在此过程中,硫原子转化为半胱氨酸(Cys)。转硫反应主要发生在肝和肾脏,Hcy与丝氨酸在胱硫醚合成酶(CBS)的催化下,以VitB6为辅酶,缩合成胱硫醚,继而在γ-胱硫醚裂解酶催化作用下,仍以5-磷酸吡哆醛为辅酶,胱硫醚裂解为半胱氨酸和牛磺酸,半胱氨酸硫原子经过大量酶促反应氧化为硫酸盐,其中大部分作为尿无机盐从体内排泄。这是一条不可逆代谢途径。(3)甲基化替代途径。甜菜碱可提供1个甲基与Hcy反应,再甲基化为甲硫氨酸。(4)Hcy直接释放到细胞外液,在体内发挥各种生理作用。
在第1种情形下,包含辅酶甲钴胺的甲硫氨酸合酶(MS)催化5-甲基四氢叶酸到Hcy的转甲基反应,生成甲硫氨酸和四氢叶酸(THF)。这个反应发生在除红细胞外的所有细胞,它包含了中间产物甲钴胺结合酶(methylCbl)[6]。在这点上,Hcy代谢与细胞内的叶酸循环紧密联系。实际上,甲硫氨酸合酶(MS)是唯一的转化叶酸和5-甲基四氢叶酸为四氢叶酸的酶,而THF对体内各种细胞功能有重要的支持作用。这些包括细胞必须摄取叶酸的谷氨酸聚合反应。THF经过丝氨酸羟甲基转移酶(SHMT)和辅因子丝氨酸和PLP催化作用进一步转换为5,10-亚甲基四氢叶酸(5,10-methyleneTHF)。在亚甲基四氢叶酸还原酶和辅助因子FADH 2(FAD是维生素B2的活性形式)的还原作用下,5,10-亚甲基四氢叶酸被还原为5-甲基四氢叶酸,参与Hcy的再甲基化(见图1)[7]。由于叶酸是以5-甲基四氢叶酸形式出现在在循环中,并且MTHFR所催化的反应不可逆,为了产生THF和其它的活性叶酸(嘌呤和嘧啶代谢所需要的),进入细胞的叶酸应该通过MS催化的反应传递;而MS是依赖甲钴胺的催化反应,所以甲钴胺缺乏可能会干预细胞内叶酸循环,导致5-甲基四氢叶酸的积聚和其它叶酸衍生物的缺失[8]。甜菜碱是胆碱氧化的一个中间产物,甜菜碱依赖的再甲基化过程需在甜菜碱Hcy甲基转移酶(BHMT)的作用下,甜菜碱作为甲基供体,使Hcy再循环为甲硫氨酸和非叶酸甲基供体。BHMT主要在肝脏和肾脏中表达。
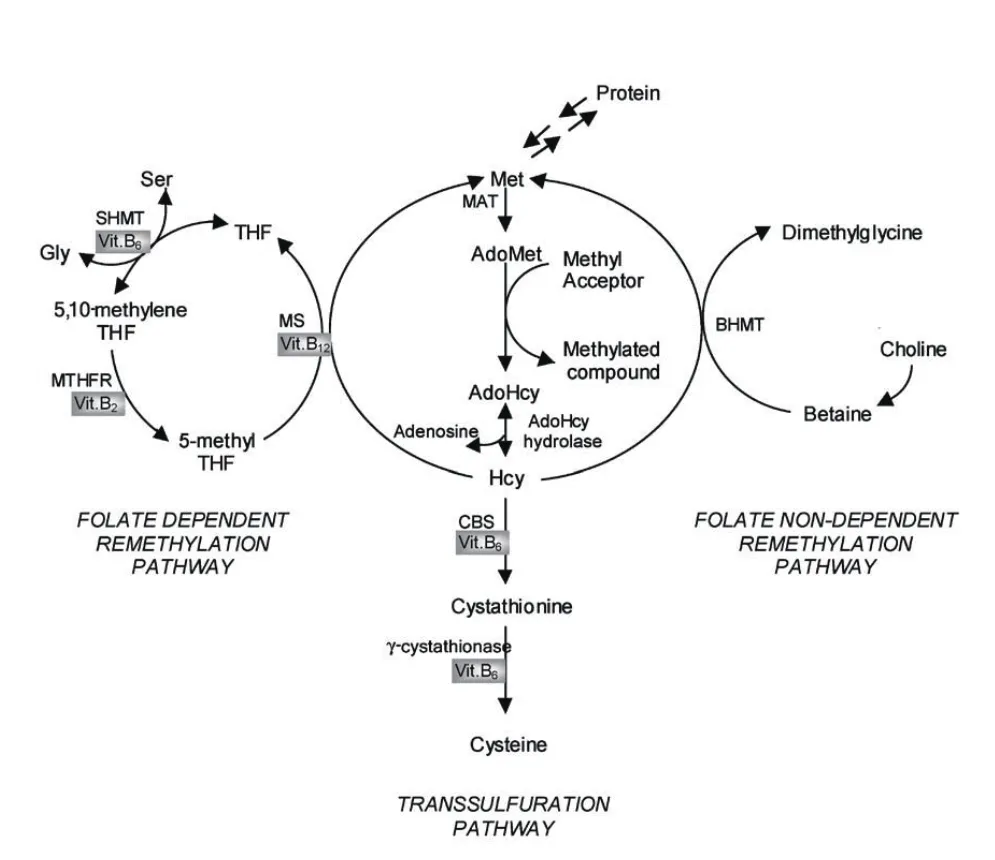
图1 同型半胱氨酸代谢和甲硫氨酸循环示意图
2 Hcy 浓度的调节
Hcy代谢的调控包括上调和下调参与再甲基化和转硫化途径中的各种酶。甲硫氨酸(Met)抑制MS和BHMT;AdoMet 抑制BHMT,同时也抑制肝MAT和MTHFR;但是它也可以活化CBS和肝MAT,间接辅助甘氨酸N-甲基转移酶作用[3]。甘氨酸N-甲基转移酶(GNMT)催化依赖S-腺苷基蛋氨酸的甘氨酸甲基化形成N-甲基甘氨酸(肌氨酸),而5-甲基四氢叶酸可以抑制此种作用[9]。当AdoMet浓度升高时,MTHFR的催化作用被抑制,因此,5-甲基四氢叶酸的浓度将会减小,对GNMT的抑制作用也因之减少。GNMT将转化过多甲基团为非毒性产物(肌氨酸),结果AdoMet浓度下降到正常。因此,增加AdoMet浓度会抑制叶酸依赖性和非依赖性的Hcy再甲基化,会促使Hcy通过转硫化途径的分解代谢和通过GNMT产生肌氨酸。
如上所述,细胞内的Hcy浓度应该在严格控制下,由于AdoHcy可以影响转甲基反应,因此,体内它的浓度增加应该被避免;AdoHcy水解酶反应的动态特征,使细胞内Hcy浓度可以保持在严格范围内。通过叶酸依赖的再甲基化可以保证细胞内最佳Hcy浓度,一旦超过这种能力,Hcy将会主动导出细胞外(见图2)。Blom等[10]假设可能存在降低Hcy浓度的运载体进行穿膜作用,并且这种作用可能被AdoMet、AdoHcy和Hcy严格控制。因此,血浆Hcy浓度密切的反应了细胞内Hcy和AdoHcy浓度,同时也反应了代谢途径中各个反应环节的完整性。
当Hcy进入血液后,由于它的巯基在氧化环境下的高反应性,它将很快氧化和/或经历一系列二硫化物取代反应,可用的取代基可以是巯基和二硫化物混合物,也可以是Hcy、Cys、谷胱甘肽、γ-谷氨酰半胱氨酸、半胱氨酰甘氨酸或蛋白质(主要白蛋白)(见图2)。因此,在正常血浆中的Hcy,仅仅1%~2%是以游离Hcy形式存在(具有1个自由的巯基团),剩余的98%是以氧化的Hcy形式存在,如以二氧化硫形式存在[11-14];并且这些中间大约75%与蛋白结合,剩余部分以非结合蛋白的二氧化硫形式存在。tHcy主要指经过定量的双硫键还原裂解后所出现的Hcy总量(见图2),目前临床和科研上被广泛应用。
肝肾是完成Hcy分解代谢和排出的主要器官,这些器官表达高浓度的CBS和BHMT。除此之外肝脏特异性MAT也呈高水平表达,且与Met有相对高Km。实际上,虽然在几种组织CBS是可以测量的,但是在肝肾外它的活性太低而不足以显著促进Hcy代谢[15]。相应地,肝肾细胞的摄取可能要求能够识别Hcy不同存在形式的载体、通道或者受体(见图2)。肝脏代谢占主要部分的Hcy结合蛋白,而剩余部分低分子量Hcy混合物很可能通过肾脏代谢,它们通过肾脏细胞时,由于细胞内的还原条件,Hcy将被释放和代谢。Refsum等[16]实验计算血浆中大约有70% Hcy被肾脏代谢,强调了肾功能对维持血循环中Hcy正常浓度的重要性。

图2 同型半胱氨酸代谢的调节—细胞摄取、输出和氧化[10]
3 高 Hcy 血症的诊断和影响因素
3.1 高 Hcy 血症(Hyperhomocysteinaemia,HHcy)HHcy指血循环中总Hcy(tHcy)浓度增加的状况。血浆tHcy的测量有2种,即空腹状态和半胱氨酸负荷后检查(MLT);这2种检查都被应用于生化诊断检测。空腹状态下血浆tHcy测定,正常参考范围在假定健康人统计值的2.5%~97.5%区间[17];MLT包含2种检测,一个是空腹血浆tHcy测定,另一个是经过摄取标准(100 mg/kg)(非生理量)半胱氨酸2~6 h后tHcy的重复测定。这种检测起初用来检测诊断CBS缺陷的杂合现象,现在常用来鉴别诊断空腹血浆tHcy可能正常,但是Hcy代谢轻度损伤的个体。MLT一直被认为是比较安全的检查[18],但有报道1例MLT检测患者死亡可能与使用半胱氨酸负荷剂量过大有关[19]。另外,MLT的临床价值也一直被怀疑和不确定[20-21]。尽管如此,人们仍然认为异常的MLT结果还是可以反映转硫化途径的异常,而空腹状态下高Hcy血症反映Hcy再甲基化的缺陷。
3.2 HHcy血症影响因素 如上文所述,如果体内细胞代谢Hcy的能力超过正常,Hcy即会输出到细胞外,直到细胞内Hcy的水平正常时才会停止。假如有任何因素影响细胞内Hcy代谢,细胞将不能达到理想浓度,那么Hcy将持续输出,最终导致Hcy积聚在血液中,引起HHcy血症。我们根据基因背景的缺失和存在,将这些影响因素进行分类如下。
3.2.1 Hcy血症的非基因调节
(1)营养因素:血浆tHcy的影响因素显然包括不适当的维生素B族浓度,维生素B族在Hcy代谢中的作用非常重要,因为B族维生素担当Hcy代谢酶的辅因子或底物。如图1所示,PLP是维生素B6的活性形式,是反应酶CBS、γ-胱硫醚酶和SHMT的辅因子;FAD是维生素B2的活性形式,是酶MTHFR和MS的辅因子,FMN是维生素B2的另一种活性形式,是MS的辅因子;甲钴胺是维生素B12的活性形式,是MS酶的辅因子;叶酸(维生素9)在Hcy再甲基化的叶酸作用途径中是协同底物。维生素B族缺陷可能是轻中度高Hcy血症的最普遍的原因[22]。维生素B12作为蛋氨酸合成酶的辅酶,它的浓度降低引起Hcy形成蛋氨酸受阻而致Hcy水平升高[23]。维生素B6是胱硫醚β合成酶及胱硫醚酶的辅酶,它的缺乏致酶活性下降引起Hcy代谢障碍。叶酸缺乏导致Hcy再甲基化障碍,并且影响亚甲基四氢叶酸还原酶的活性,进一步影响甲基四氢叶酸的生成,导致Hcy水平明显升高。血浆tHcy浓度与血浆中叶酸、维生素B12和维生素B6呈负相关,并且与这些维生素的摄取也呈负相关[24]。其中呈现最一致联系的是与叶酸的低摄取和叶酸的低血浆浓度负相关[25-28]。HHcy血症的当前治疗证实作为血浆tHcy非基因影响因素的维生素作用,它常可以减少空腹高Hcy血症的25%;维生素B12可以减少空腹tHcy浓度的7%,而维生素B6对空腹tHcy浓度却没有作用[29],但是它却可以减少MLT tHcy浓度20%~30%[30]。有研究显示,在维生素B2与tHcy浓度之间也有一定的负相关关系[31],然而这种联系被限定在低血浆浓度的对象并且基因型是MTHFR 677 TT[32]。
(2)肾功能:血清肌酸酐一直是影响空腹tHcy浓度的决定因素之一,反映了在这些浓度下肾功能的作用[33-35]。肌酸合成是AdoMet依赖性的转甲基反应,这也说明了在正常人群中,肌酐与空腹tHcy浓度之间的直接联系[36-37],肾衰竭常伴随着tHcy浓度的升高[34]。有研究报道,肾小球滤过率与血浆tHcy浓度呈显著负相关[33,35]。相应地,半胱氨酸蛋白酶抑制剂C作为肾小球滤过率的标志物和tHcy浓度的主要影响因素,在健康人群和肾衰病人中,它的浓度也与血浆tHcy浓度有相关性[38-39]。然而在这些患者中,高Hcy的根本原因仍不清楚。目前认为,受损的肾脏清除能力、代谢物增加或者叶酸代谢缺陷都不能解释肾功能衰竭时的HHcy血症,唯一可能的原因是肾功能下降和肾外Hcy的代谢[40]。
(3)性别:基于大规模人群研究明确显示,血浆高tHcy浓度与男性人群密切相关,男性比女性人群更高[41-43]。Fukagawa等[44]的研究进一步表明,绝经前女性再甲基化和转甲基化率都高于男性,这可能是由于MS和BHMT酶活性存在性别相关的差异;而研究认为在绝经期妇女tHcy浓度的增加可能由于类固醇激素的影响[45-48]。雌激素增加甜菜碱Hcy转甲基酶活性,从而促进Hcy代谢,降低其血浆浓度[49]。男性Hcy水平较高还与男性肌酐浓度和肌肉含量较高有关[50];由于肌酐与肌肉质量有关,男女性血液循环中肌酐有差异性,这可能会部分解释tHcy浓度的性别相关性差异[51]。男性和女性人群中维生素B营养状况不同也可以促使这种差异更显著。
(4)年龄:血浆Hcy浓度随着增龄会逐渐增加[52-53],从青少年阶段到老年阶段tHcy血浆浓度大约增加1倍[54]。目前认为原因可能与老年人维生素B6、维生素B12摄取水平下降,出现不同程度的肾功能减退和胱硫醚酶活性降低,导致tHcy血浆浓度增加[55]。
(5)其它因素:妊娠期间血浆tHcy浓度显著减少[56]。吸烟、咖啡、缺乏锻炼和饮酒都可以增加血浆tHcy浓度。令人感兴趣的是,适度饮酒者比不饮酒者血浆tHcy浓度更低[57]。有些药物如氨甲喋吟、卡马西平、苯妥英钠通过干扰叶酸或含硫氨基酸代谢,引起一过性血浆Hcy升高。传统的心血管危险因素如血脂和血压也与血浆tHcy浓度呈正相关[58]。某些疾病如乳腺癌、胰腺癌、卵巢癌,急性淋巴细胞白血病、糖尿病时可以观察到Hcy水平升高。
3.2.2 Hcy血症的基因调节
(1)CBS缺乏:由于CBS缺陷引起的HHcy血症和高胱氨酸尿症(homocystinuria)是一种常见的先天性代谢缺陷性疾病,我们以高胱氨酸尿症说明Hcy的调节,此病常有典型的临床表现和病理特征[59]。高胱氨酸尿症存在2种主要表现型,一种是轻度的PLP易感型,另一种为更严重的非易感型;影响到4种组织器官主要包括:眼睛、骨骼、中枢神经系统和血液系统[60],然而这些联合损伤的潜在病理生理机制并没有完全清楚。此病最主要和最频繁的死亡原因是血栓栓塞,血管闭塞可以发生在任何血管和任何年龄[3]。生化检测发现不管在血浆还是尿中,患者具有高浓度的tHcy;并且血浆中有高浓度的Met。早期降低tHcy治疗包括单独补充维生素B6或者与叶酸和甜菜碱联合补充,尽管并不是十分理想的生化治疗,但还是可以显著降低威胁生命的心血管危险因素[61-62]。
最新研究发现,CBS基因有100多种突变,被绘制在21 q 22.3;大多数的变异是错义突变,I 278 T和G 307 S是最普遍的变异。此外,还发现了剪接变异,插入突变和中间缺失等基因变异[63-65]。人类CBS是同源四聚体,结合了2种辅因子(PLP和血红素)[66]。PLP参与基本结构反应,血红素可能作为氧化-还原的传感器[67]。每个CBS单体有551个氨基酸残基,结合2个底物(Hcy和丝氨酸),进而被AdoMet所调节。
(2)MTHFR缺陷:高胱氨酸尿症也可以由于MTHFR基因变异而导致[68-70]。这种罕见的常染色体异常引起高Hcy血症和具有低或正常的蛋氨酸浓度的高胱氨酸尿症,其原因为Hcy再甲基化为Met能力下降。临床症状包括血管和神经上的异常;此种患者有较低的酶活性,并发现与出现症状时的年龄相关[71]。迄今为止,研究报道已发现33种MTHFR基因变异现象,这些变异可以引起严重的高Hcy血症[8]。
(3)功能性MS缺陷:蛋氨酸合酶(MS)(CblG)和还原酶(MSR)(CblE)缺陷是一种罕见的情况,细胞内钴胺族(Cbl)代谢紊乱间接影响了MS活性,也导致高胱氨酸尿症的发生[8]。钴胺族经过在细胞溶胶中第一步共同反应后,它转化为活性形式:腺钴胺和甲钴胺。其中腺钴胺形成在线粒体,是甲基丙二酰辅酶A变位酶的辅助因子,可以转化甲基丙二酰辅酶A为琥珀酰CoA。甲钴胺通过MS的催化作用在胞质溶胶生成,5-甲基THF提供甲基团给酶结合的钴胺,通过转甲基作用生成甲钴胺,随后甲基团从甲钴胺传递给Hcy生成Met[72]。MS的活性又要依赖MSR催化钴铵(Ⅱ)生成钴铵(Ⅰ)的复合作用来完成[73]。CblG可以引起编码MS的MTR基因变异而导致酶作用异常;CblE引起编码MSR的MTRR 基因变异,两者都导致不能产生MS的辅因子甲钴胺[74]。这两种病因引起的疾病表现出同样的生物化学检测结果(无甲基丙二酸的高胱氨酸尿症)和临床特征,临床特征包含了大量的血液和神经精神系统的异常。两种病因只能通过体细胞互补分析才能鉴别它们之间的差异。
综上所述,Hcy的体内代谢是人体非常重要的代谢途径,其代谢途径中的异常会导致体内多种疾病的发生;HHcy的调节有基因和非基因调节因素,如果调节因素异常将会导致高同型半胱氨酸血症,并引起某些相关临床疾病的发生,因此对于Hcy的代谢及调节因素的研究具有非常重要的意义。
[1] Finkelstein JD,Martin JJ.Methionine metabolism in mammals[J].Adaptation to methionine excess.J Biol Chem,1986,261:1582-1587.
[2] Akerman K,Karkola K,Kajander O.Methionine adenosyltransferase activity in cultured cells and in human tissues[J].Biochim Biophys Acta,1991,1097:140-144.
[3] Mudd SH,Levy HL,Krauss JP.Disorders of transsulfuration.In:Scriver CR,Beaudet AL,Sly WS,Valle D,eds;Childs B,Kinzler KW,Vogelstein B,assor.eds.The Metabolic and Molecular Bases of Inherited Disease[M].8 th edn,vol.2.New York:McGraw-Hill,2011:2007-2056.
[4] Scott JM,Weir DG,Molloy A,et al.Folic acid metabolism and mechanisms of neural tube defects[J].Ciba Found Symp,2010,181:180-191.
[5] Friso S,Choi SW,Girelli D,et al.Acommon mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status[J].Proc Natl Acad Sci USA,2012,99:5606-5611.
[6] Mudd SH,Skovby F,Levy HL,et al.The natural history of homocystinuria due to cystathionine beta-synthase deficiency[J].Am J Hum Genet,1985,37:1-31.
[7] Finkelstein JD.The metabolism of homocysteine:pathways and regulation [J].Eur J Pediatr,2012,57:S 40-44.
[8] Rosenblatt DS,Fenton WA.Inherited disorders of folate and cobalamin transport and metabolism.In:Scriver CR,Beaudet AL,Sly WS,Valle D,eds;Childs B,Kinzler KW,Vogelstein B,assoc.eds.The Metabolic and Molecular Basies of Inherited Disease[M].8 th edn,vol.3.New York:McGraw Hill,2011,3897-3933.
[9] Stipanuk MH.Sulphur amino acid metabolism:pathways for production and removal of homocysteine and cysteine[J].Annu Rev Nut,2004,24:539-577.
[10] Blom HJ.Consequences of homocysteine export and oxidation in the vascular system[J].Semin Thromb Hemost,2010,26:227-232.
[11] Andersson A,Lindgren A,Hultberg B.Effect of thiol oxidation and thiol export from erythrocytes on determination of redox status of homocysteine and other thiols in plasma from healthy subjects and patients with cerebral infarction[J].Clin Chem,1995,41:361-366.
[12] Hultberg B,Andersson A,Arnadottir M.Reduced,free and total fractions of homocysteine and other thiol compounds in plasma from patients with renal failure[J].Nephron,1995,70:62-67.
[13] Mansoor MA,Bergmark C,Svardal AM,et al.Redox status and protein binding of plasma homocysteine and other aminothiols in patients with early-onset peripheral vascular disease.Homocysteine and peripheral vascular disease[J].Arterioscler Thromb Vasc Biol,1995,15:232-240.
[14] Ueland PM.Homocysteine species as components of plasma redox thiol status[J].Clin Chem,1995,41:340-342.
[15] Vander Molen EF,Hiipakka MJ,van Lith-Zanders H,et al.Homocysteine metabolism in endothelial cells of a patient homozygous for cystathionine beta-synthase (CS) deficiency [J].Thromb Haemost,2012,8:827-833.
[16] Refsum H,Guttormsen AB,Fiskerstrand T,et al.Hyperhomocysteinaemia in terms of steady-state kinetics[J].Eur J Pediatr,1998,157(Supplement 2):S 45-49.
[17] Refsum H,Smith AD,Ueland PM,et al.Facts and recommendations about total homocysteine determinations:an expert opinion[J].Clin Chem,2014,50:3-32.
[18] Krupkova-Meixnerova L,Vesela K,Vitova A,et al.Methionineloading test:evaluation of adverse effects and safety in an epidemiological study[J].Clin Nutr,2012,21:151-156.
[19] Cottington EM,LaMantia C,Stabler SP,et al.Adverse event associated with methionine loading test:a case report[J].Arterioscler Thromb Vasc Biol,2002,22:1046-1050.
[20] Fokkema MR,Dijck-Brouwer DA,van Doormaal JJ,et al.Low diagnostic value of fasting and post-methionine load homocysteine tests.Astudy in Dutch subjects with homocysteine test indications[J].Clin Chim Acta,2003,331:153-157.
[21] Fokkema MR,Gilissen MF,Van Doormaal JJ,et al.Fasting vs nonfasting plasma homocysteine concentrations for diagnosis of Hyperhomocysteinaemia[J].Clin Chem,2003,49:818-821.
[22] Haynes WG.Hyperhomocysteinaemia,vascular function and atherosclerosis:effects of vitamins[J].Cardiovasc Drugs Ther,2012,16:391-399.
[23] Tucker KL,Hannan MT,Qiao N,et al.Low plasma vitamin B 12 is associated with lower BMD:the Framingham Osteoporosis Study[J].J Bone Miner Res,2005,20:152-158.
[24] Graham IM,O’Callaghan P.Vitamins,homocysteine and cardiovascular risk[J].Cardiovasc Drugs Ther,2002,16:383-389.
[25] Ganji V,Kafai MR.Demographic,health,lifestyle,and blood vitamin determinants of serum total homocysteine concentrations in the third National Health and Nutrition Examination Survey[J].Am J Clin Nutr,2003,77(4):826-833.
[26] Jacques PF,Bostom AG,Wilson PW,et al.Determinants of plasma total homocysteine concentration in the Framingham Offspring cohort[J].Am J Clin Nutr,2001,73:613-621.
[27] Nygard O,Refsum H,Ueland PM,et al.Major lifestyle determinants of plasma total homocysteine distribution:the Hordaland Homocysteine Study[J].Am J Clin Nutr,1998,67:263-270.
[28] Selhub J,Jacques PF,Wilson PW,et al.Vitamin status and intake as primary determinants of homocysteinemia in an elderly population[J].JAMA,1993,270:2693-2698.
[29] Homocysteine Lowering Trialists’ Collaboration.Lowering blood homocysteine with folic acid based supplements:meta analysis of randomised trials[J].BMJ,1998,316:894-898.
[30] Bostom AG,Gohh RY,Beaulieu AJ,et al.Treatment of Hyperhomocysteinaemia in renal transplant recipients.A randomized,placebo-controlled trial[J].Ann Intern Med,2012,127:1089-1092.
[31] Morris MS,Jacques PF,Selhub J.Relation between homocysteine and B vitamin status indicators and bone mineral density in older Americano[J].Bone,2005,37:234-242.
[32] Jacques PF,Kalmbach R,Bagley PJ,et al.The relationship between riboflavin and plasma total homocysteine in the Framingham Offspring cohort is influenced by folate status and the C 677 T transition in the methylenetetrahydrofolate reductase gene[J].J Nutr,2002,132:283-288.
[33] Arnadottir M,Hultberg B,Nilsson-Ehle P,et al.The effect of reduced glomerular filtration rate on plasma total homocysteine concentration[J].Scand J Clin Lab Invest,2011,56:41-46.
[34] Bostom AG,Culleton BF.Hyperhomocysteinaemia in chronic renal disease[J].J Am Soc Nephrol,1999,10:891-900.
[35] Wollesen F,Brattstrom L,Refsum H,et al.Plasma total homocysteine and cysteine in relation to glomerular filtration rate in diabetes mellitus[J].Kidney Int,1999,55:1028-1035.
[36] Brattstrom L,Lindgren A,Israelsson B,et al.Homocysteine and cysteine:determinants of plasma levels in middle-aged and elderly subjects[J].J Intern Med,1994,236:633-641.
[37] Wu LL,Wu J,Hunt SC,et al.Plasma homocyst(e)ine as a risk factor for early familial coronary artery disease[J].Clin Chem,1994,40:552-561.
[38] Amouzou EK,Chabi NW,Adjalla CE,et al.High prevalence of hyperhomocysteinemia related to folate deficiency and the 677 C→T mutation of the gene encoding methylenetetrahydrofolate reductase in coastal West Africa[J].Am J Clin Nutr,2004,79:619-624.
[39] Anwar A,Gu´eant JL,Abdelmouttaleb I,et al.Hyperhomocysteinemia is related to residual glomerular filtration and folate,but not to methylenetetrahydrofolate reductase and methionine synthase polymorphisms,in supplemented end-stage renal disease patients undergoing hemodialysis[J].Clin Chem Lab Med,2011,39:747-752.
[40] Perna AF,Ingrosso D,Satta E,et al.Homocysteine metabolism in renal failure[J].Curr Opin Clin Nutr Metab Care,2014,7:53-57.
[41] Ravaglia G,Forti P,Maioli F,et al.Folate,but not homocysteine,predicts the risk of fracture in elderly persons[J].J Gerontol A Biol Sci Med Sci,2005,60:1458-1462.
[42] Jacques PF,Rosenberg IH,Rogers G,et al.Serum total homocysteine concentrations in adolescent and adult Americans:results from the third National Health and Nutrition Examination Survey[J].Am J Clin Nutr,1999,69:482-489.
[43] Nygard O,Vollset SE,Refsum H,et al.Total plasma homocysteine and cardiovascular risk profile.The HordalandHomocysteine Study[J].JAMA,1995,274:1526-1533.
[44] Fukagawa NK,Martin JM,Wurthmann A,et al.Sex-related differences in methionine metabolism and plasma homocysteine concentrations[J].Am J Clin Nutr,2000,72:22-29.
[45] Brattstrom LE,Hultberg BL,Hardebo JE.Folic acid responsive postmenopausal homocysteinemia [J].Metabolism,2012,34:1073-1077.
[46] Giltay EJ,Hoogeveen EK,Elbers JM.Effects of sex steroids on plasma total homocysteine levels:a study in transsexual males and females[J].J Clin Endocrinol Metab,1998,83:550-553.
[47] Bozkurt N,Erdem M,Yilmaz E,et al.The relationship of homocysteine,B 12 and folic acid with the bone mineral density of the femur and lumbar spine in Turkish postmenopausal women[J].Arch Gynecol Obstet,2009,280:381-387.
[48] Biagini MR,Tozzi A,Bongini E,et al.Associationof plasma homocysteine with bone mineral density in postmenopausal women withosteoporosis or osteopenia affected by primary biliary cirrhosis[J].J Clin Gastroenterol,2007,41:635.
[49] Finkelstein JD.Metabolic regulatory properties of S-adenosylmethionine and Sadenosylhomocysteine[J].Clin Chem Lab Med,2007,45:1694-1699.
[50] Chauveau P,Chadefaux B,Coude M,et al.Hyperhomocysteinemia,a risk factor for atheroselerosis in chronic uremic patients[J].Kidney Int,1993,41:s 72-s 77.
[51] Lussier-Cacan S,Xhignesse M,Piolot A,et al.Plasma total homocysteine in healthy subjects:sex-specific relation with biological traits[J].Am J Clin Nutr,1996,64:587-593.
[52] Sunghoon K.Molecular biology of aging[J].Arch Surg,2003,138:1051-1054.
[53] Krumdieck Carlos L,Prince Charles W.Mechanisms of homocysteine toxicity on connective tissues:implications for the morbidity of aging[J].J Nutr,2000,130:365 S-368 S.
[54] Refsum H,Fredriksen A,Meyer K,et al.Birth prevalence of homocystinuria[J].J Pediatr,2014,144:830-822.
[55] Perez FP,Ilie JI,Zhou XM.Pathomolecular effects of homocysteine on the aging process:A new theory of aging[J].Medical Hypotheses,2007,69:149-160.
[56] Andersson A.Decreased serum homocysteine in pregnancy[J].European Journal of Clinical Chemistry and Clinical Biochemistry,1992,30:377-379.
[57] Hultherg B,Berglund M,Andersson A,et al.Elevated plasma homocysteinein alcoholics[J].Alcohol Clin Exp Res,1993,17(3):687-689.
[58] Lievers KJ,Kluijtmans LA,Blom HJ.Genetics of hyperhomocysteinaemia in cardiovascular disease[J].Ann Clin Biochem,2013,40:46-59.
[59] Refsum H,Smith AD,Ueland PM,et al.Facts and recommendations about total homocysteine determinations:an expert opinion[J].Clin Chem,2014,50:3-32.
[60] Sokolova J,Janosikova B,Terwilliger JD,et al.Cystathionine betasynthase deficiency in Central Europe:discrepancy between biochemical and molecular genetic screening for homocystinuric alleles[J].Hum Mutat,2001,18:548-549.
[61] Yap S.Classical homocystinuria:vascular risk and its prevention[J].J Inherit Metab Dis,2003,26:259-265.
[62] Yap S,Boers GH,Wilcken B,et al.Vascular outcome in patients with homocystinuria due to cystathionine beta-synthase deficiency treated chronically:a multicenter observational study[J].Arterioscler Thromb Vasc Biol,2001,21:2080-2085.
[63] Meier M,Oliveriusova J,Kraus JP,et al.Structural insights into mutations of cystathionine beta-synthase[J].Biochim Biophys Acta,2003,1647:206-213.
[64] Skovby F,Krassikoff N,Francke U.Assignment of the gene for cystathionine beta-synthase to human chromosome 21 in somatic cell hybrids[J].Hum Genet,2014,65:291-294.
[65] Munke M,Kraus JP,Ohura T,et al.The gene for cystathionine beta-synthase (CBS) maps to the subtelomeric region on human chromosome 21 q and to proximal mouse chromosome 17[J].Am J Hum Genet,1988,42:550-559.
[66] Banerjee R,Evande R,Kabil O,et al.Reaction mechanism and regulation of cystathionine beta-synthase[J].Biochim Biophys Acta,2003,1647:30-35.
[67] Kery V,Poneleit L,Meyer JD,et al.Binding of pyridoxal phosphate to the heme protein human cystathionine beta-synthase[J].Biochemistry,2012,38:2716-2724.
[68] Goyette P,Christensen B,Rosenblatt DS,et al.Severe and mild mutations in cis for the methylenetetrahydrofolate reductase (MTHFR)gene,and description of five novel mutations in MTHFR[J].Am J Hum Genet,1996,59:1268-1275.
[69] Goyette P,Frosst P,Rosenblatt DS,et al.Seven novel mutations in the methylenetetrahydrofolate reductase gene and genotype phenotype correlations in severe methylenetetrahydrofolate reductase deficiency[J].Am J Hum Genet,1995,56:1052-1059.
[70] Goyette P,Sumner JS,Milos R,et al.Human methylenetetrahydrofolate reductase:isolation of cDNA mapping and mutation identification[J].Nature Genetics,1994,7:551.
[71] Sibani S,Christensen B,O’Ferrall E,et al.Characterization of six novel mutations in the methylenetetrahydrofolate reductase (MTHFR) gene in patients with homocystinuria[J].Hum Mutat,2012,15:280-287.
[72] Watkins D,Ru M,Hwang HY,et al.Hyperhomocysteinaemia due to methionine synthase deficiency,cblG:structure of the MTR gene,genotype diversity,and recognition of a common mutation,P 1173 L[J].Am J Hum Genet,2002,71:143-153.
[73] Leclerc D,Wilson A,Dumas R,et al.Cloning and mapping of a cDNA for methionine synthase reductase,a flavoprotein defective in patients with homocystinuria[J].Proc Natl Acad SciUSA,2011,95:3059-3064.
[74] Wilson A,Leclerc D,Rosenblatt DS,et al.Molecular basis for methionine synthase reductase deficiency in patients belonging to the cblE complementation group of disorders in folate/cobalamin metabolism[J].Hum Mol Genet,1999,8:2009-2016.
