分子内氢键对屏蔽酚抗氧化活性的影响
2014-12-31段庆华武志强王立华李新华
苏 朔,龙 军,段庆华,周 涵,武志强,赵 毅,王立华,李新华
(中国石化 石油化工科学研究院,北京 100083)
屏蔽酚是指在酚羟基的邻位存在叔丁基等较大取代基的酚类化合物。目前,屏蔽酚抗氧剂已广泛应用于人体 健 康[1]、 食 品[2]、 聚 合 物[3]、 润 滑 油[4]等诸多领域。由于其具有低毒、无磷、无灰等优点,在高档润滑油中拥有广阔的应用前景。
在润滑油体系中,屏蔽酚主要通过捕获烃分子氧化过程中形成的最初始烷基过氧自由基,从而起到抗氧化作用,如图1所示[4]。氢转移反应生成的苯氧自由基不仅受到邻位取代基团空间位阻效应的保护[5],还可以通过共振结构继续捕获过氧自由基,从而抑制自由基链传递。过氧自由基除了与屏蔽酚反应,也可能继续与烃分子反应;2个反应均为氢转移反应,也是一对竞争反应。因此,屏蔽酚的供氢能力是影响其抗氧化活性的一项重要因素,供氢能力越强,其抗氧化活性将越高。屏蔽酚的O—H键解离能EBD作为表征O—H共价键强度的一项重要参数[6-7],可以较准确地反映酚羟基供氢能力的强弱。因此,EBD常被用于评价屏蔽酚的化学反应活性及热力学性质[8-9]。EBD越小,表明供氢能力越强,抗氧化活性越高。
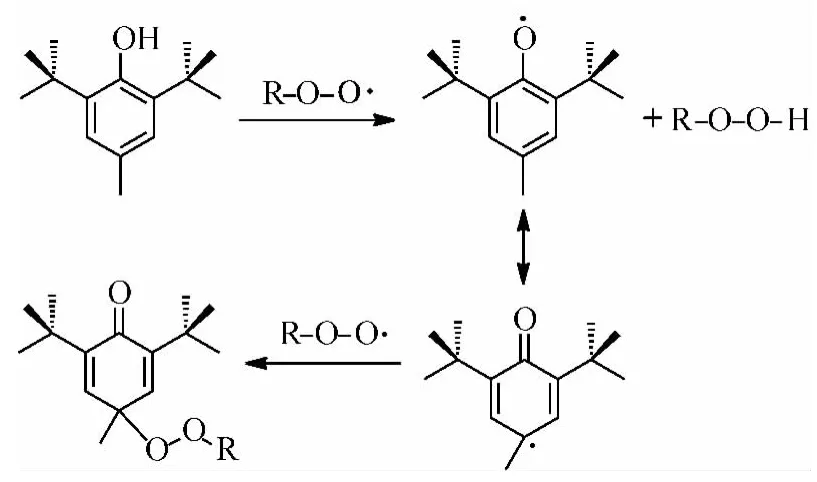
图1 屏蔽酚与过氧自由基的反应Fig.1 The reactions between hindered phenol and peroxide radical
Lucarini等[10-11]采用电子自旋共振(EPR)方法研究屏蔽酚的氢转移反应平衡常数,测定了屏蔽单酚、邻位桥联屏蔽双酚以及对位桥联屏蔽双酚的EBD,发现分子内氢键影响屏蔽酚的EBD。Tomiyama等[8]采用量子化学从头计算方法研究了单取代的酚分子与过氧自由基之间的氢转移反应,发现氢转移反应的能垒Ea与酚的EBD之间存在很高的相关性。但是,对于屏蔽酚的EBD与其在润滑油基础油中的抗氧化活性之间存在怎样的关系,尚未见报道,对于影响屏蔽酚EBD的结构因素,现有认识也不全面。笔者结合密度泛函理论(DFT)方法和加压差示扫描量热(PDSC)手段,考察了不同结构屏蔽酚的EBD与其在基础油中抗氧化活性的关系,并重点研究了分子内氢键和分子内旋转对邻位桥联屏蔽双酚EBD的影响。
1 实验部分
1.1 屏蔽酚EBD的计算方法
考虑到相对分子质量较小的屏蔽酚在较高温度条件下会产生较严重的热挥发损失,因此,在本研究中重点选取了5个相对分子质量较大的屏蔽酚作为模型化合物,其分子结构如图2所示。
采用分子模拟软件 Materials Studio 6.0中的DMol3量子力学模块,对屏蔽酚分子和相关自由基的几何结构进行优化,计算EBD的公式如式(1)所示。

式 (1)中,E(Ar—O·) 、E(H·)和E(Ar—O—H)分别代表苯氧自由基、氢自由基和屏蔽酚分子的能量,kJ/mol。涉及到的能量计算均考虑了零点振动能(EZPV)校正。采用过渡态理论计算邻位桥联屏蔽双酚的内旋转能垒,其中,过渡态的搜索采用完全线性同步和二次同步变换(Complete LST/QST)方法。采用基于广义梯度近似(GGA)的PW91泛函方法,在DNP基组(双数值轨道基组+p轨道极化函数)水平上进行全电子计算。能量、受力和位移的收敛标准分别为2×10-5Ha、0.04Ha/nm和5×10-4nm,自洽场(SCF)迭代收敛的阈值设为1×10-5Ha。
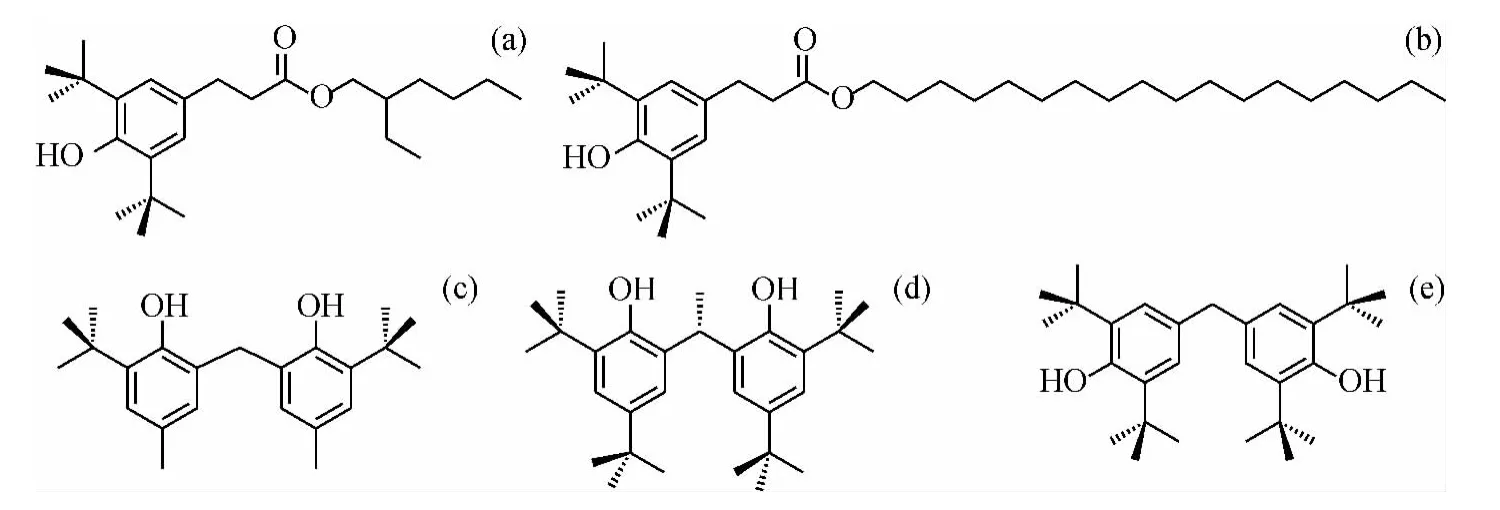
图2 5种屏蔽酚的分子结构Fig.2 The molecular structures of five hindered phenols
1.2 原料和试剂
中国石化上海高桥石化II类加氢基础油,其主要物化性质如表1所示。

表1 II类加氢基础油的主要物化性质Table 1 Physicochemical properties of II hydrogenated base oil
5种屏蔽酚化合物,HP-1、HP-2、HP-3、HP-4、HP-5,纯度均大于98.0%。HP-1、HP-5,中国石化石油化工科学研究院产品;HP-2,南京华立明科商贸有限公司产品;HP-3,梯希爱(上海)化成工业发展有限公司产品;HP-4,美国 ALDRICH Chemical公司产品。
1.3 仪器和测定方法
采用美国Nicolet公司Nicolet560型傅里叶变换红外光谱仪测定样品的FT-IR谱,分辨率4cm-1,范围400~4000cm-1,扫描次数16次,扫描速率0.63s。
采用美国TA仪器公司TA5000-DSC2910型差热扫描仪进行加压差示扫描量热实验(PDSC),样品质量(3.0±0.1)mg,O2压力0.5MPa、流速100mL/min,升温速率100℃/min。采用恒温法时温度为210℃。测量试样自恒温开始至出现氧化放热峰的时间,取2次实验数据的平均值,即为氧化诱导期(OITPDSC)。OITPDSC越大,表明屏蔽酚在基础油中的抗氧化活性越高。为准确考察屏蔽酚抗氧化官能团酚羟基的抗氧化活性,均按相等的酚羟基质量摩尔浓度(5.0×10-5mol/g)在基础油中添加屏蔽酚。
2 结果与讨论
2.1 不同分子结构屏蔽酚的最低能量构象的EBD与其抗氧化活性的关系
分子的最低能量构象代表了其最稳定的分子构象,因此EBD的计算首先考虑最低能量构象的EBD值。表2为不同分子结构屏蔽酚的最低能量构象EBD与其OITPDSC数据。出于对计算精度的考虑,计算了最典型屏蔽酚BHT分子的EBD,可以看出PW91/DNP水平上的DFT计算值略高于实验值,偏差较小,精度较可靠。另外,BHT分子较小,其高温热挥发损失较大,故不考虑BHT的PDSC数据。

表2 不同分子结构屏蔽酚的最低能量构象EBD与其OITPDSC数据Table 2 The EBDand OITPDSCof different hindered phenols with the lowest energy conformation
由表2可知,屏蔽单酚HP-1和HP-2的EBD仅相差0.7kJ/mol,供氢能力基本一致;对位桥联屏蔽双酚 HP-5的EBD则比前2种屏蔽酚低约5kJ/mol,供氢能力更强。从PDSC氧化诱导期OITPDSC看,前2种屏蔽酚的抗氧化活性相同,而HP-5则表现出更强的抗氧化活性,这与EBD计算值所反映出的活性规律完全一致。这表明,对于屏蔽单酚和对位桥联屏蔽双酚而言,EBD越小,其抗氧化活性越高。而仅仅通过改变酚羟基对位取代基团中的碳链长度,并不能显著改善屏蔽酚的抗氧化活性。
由表2还可以看出,邻位桥联屏蔽双酚HP-3和HP-4的EBD远高于对位桥联屏蔽双酚HP-5,供氢能力相对很弱。但是,HP-4的EBD计算值比实验值高约39kJ/mol,偏差很大。而且,HP-3的氧化诱导期很长,抗氧化效果十分突出,这与其EBD较高所反映出的低抗氧化活性并不相符。
由此表明,邻位桥联屏蔽双酚的最低能量构象不是其抗氧化活性最佳构象,最低能量构象EBD不能准确表征其抗氧化活性。Amorati等[11]曾发现分子内氢键会影响邻位桥联屏蔽双酚的EBD,因此,分析了分子内氢键对邻位桥联屏蔽双酚的EBD的影响。
2.2 分子内氢键对邻位桥联屏蔽双酚EBD的影响
2.2.1 最低能量构象的分析
图3和表3分别为邻位桥联屏蔽双酚的最低能量构象和最低能量构象的结构数据。

图3 邻位桥联屏蔽双酚分子的最低能量构象Fig.3 The lowest energy molecular conformation of hindered ortho-bisphenols

表3 屏蔽双酚分子最低能量构象的结构数据Table 3 Structural data of the hindered bisphenols with the lowest energy molecular conformation
由表3可以看出,对位桥联屏蔽双酚HP-5的EBD键键长在0.0968~0.0970nm 范围,二面角C—CO—H在0°~5°范围,酚羟基基本上与苯环共面。与HP-5相比,邻位桥联屏蔽双酚 HP-3和HP-4的O—H键键长在0.0980~0.0983nm范围,显著伸长;二面角C—CO—H在20°~23°范围,也显著增大。结合图3可以看出,在HP-3和HP-4的分子最低能量构象中,酚羟基并未完全与苯环共面,并且较大幅度地伸向桥联苯环一侧。
表4列出了邻位桥联屏蔽双酚分子最低能量构象的键角和原子间距离数据。结合表4和图3可以发现,邻位桥联屏蔽双酚分子中,H26┅C9和H27┅C4的原子间距离约为0.236nm,远小于C原子和H原子的范德华半径之和0.330nm;∠O14H26C9为170°,原子O14、H26和C9近乎在一条直线上,而且原子O25、H27和C4亦是如此。因此,从形成氢键的条件推断,邻位桥联屏蔽双酚HP-3和HP-4的最低能量构象中,O—H键很可能与邻位桥联苯环的共轭π键形成π型氢键O—H┅π。受此影响,酚羟基H原子受到邻位桥联苯环共轭π电子体系的静电吸引,这是导致邻位桥联屏蔽双酚最低能量构象的EBD显著高于其他屏蔽酚的重要原因。
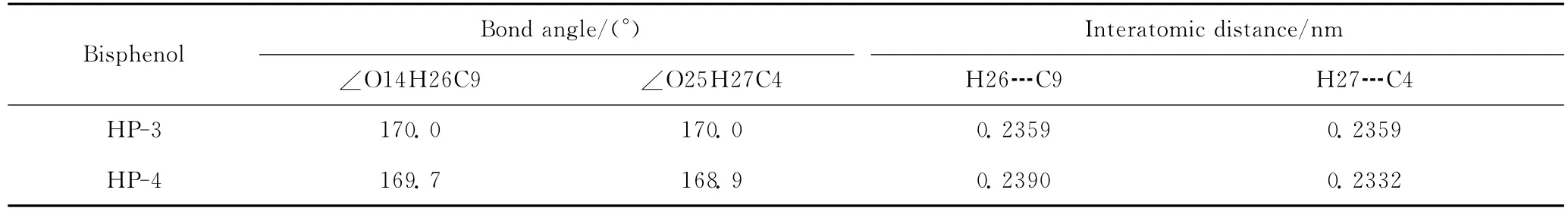
表4 邻位桥联屏蔽双酚分子最低能量构象的键角和原子间距离Table 4 The bond angle and the interatomic distance of the hindered bisphenols with the lowest energy molecular conformation
2.2.2 活性构象的分析
在优化计算分子构象过程中,发现邻位桥联屏蔽双酚HP-3和HP-4存在如图4所示的局部最低能量构象。邻位桥联屏蔽双酚局部最低能量构象的键角、原子间距离和EBD数据列于表5。
从图4和表5可以看出,HP-3和HP-4局部最低能量构象中,H27和O14的原子间距离较小,为0.189nm和0.178nm,显著低于O原子和H原子的范德华半径之和0.302nm,而且原子O25、H27和O14近乎处于同一直线上。这表明,邻位桥联屏蔽双酚HP-3和HP-4在其局部最低能量构象中会形成分子内氢键O25—H27┅O14。

图4 邻位桥联屏蔽双酚分子的活性构象Fig.4 The active conformation of hindered ortho-bisphenols

表5 邻位桥联屏蔽双酚分子活性构象的结构参数和EBD(O—H)Table 5 Structural data and EBD(O—H)of the active conformation of hindered ortho-bisphenols
由表5可以看出,O25—H27键的H27原子参与形成O—H┅O分子内氢键,对应的EBD(O25—H27)在370~383kJ/mol范围,接近最低能量构象的EBD值。O14—H26键的H26未参与形成氢键,而是O14参与形成O—H┅O分子内氢键,对应的EBD(O14—H26)在324~330kJ/mol范围,相比于最低能量构象的EBD和EBD(O25—H27),降低了约50kJ/mol。邻位桥联屏蔽双酚的局部最低能量构象,其O14—H26键均裂后所形成的苯氧自由基中心因参与形成O—H┅O分子内氢键,其稳定性得到增强,这可能是EBD(O14—H26)较低的主要原因。
由此表明,形成O—H┅O分子内氢键的局部最低能量构象是邻位桥联屏蔽双酚分子 HP-3和HP-4的抗氧化活性构象,O14—H26是其抗氧化活性官能团,拥有较强供氢能力。
2.2.3 分子内氢键的实验验证
图5为屏蔽双酚化合物的FT-IR谱。由图5可以看出,对位桥联屏蔽双酚化合物HP-5在3641cm-1位置有1个细而尖锐的吸收峰,这符合未形成氢键作用的O—H键的伸缩振动吸收峰特征。与HP-5显著不同的是,化合物HP-3在3393cm-1位置有1个较宽的吸收峰,化合物HP-4在3485cm-1位置也有1个峰形较宽的吸收峰,这是氢原子参与氢键作用的O—H键的伸缩振动吸收峰。由此表明,邻位桥联屏蔽双酚化合物HP-3和HP-4存在分子内氢键缔合作用。
分析形成氢键的O—H键伸缩振动吸收峰位置可 以 发 现,HP-3 与 HP-4 相 比, 波 数 降 低90cm-1,吸收峰显著红移,说明化合物HP-3的分子内氢键作用更强。由此推断,化合物HP-3的分子内氢键以较强的O—H┅O氢键为主,而化合物HP-4的分子内氢键则以相对弱一些的π型氢键O—H┅π为主,这进一步验证了前面的理论计算结果。
除此之外,与 HP-5相比,HP-3和 HP-4在3600~3630cm-1范围内也有1个细而尖锐的O—H键伸缩振动吸收峰,表明这2种化合物的一部分O—H键氢原子并未参与形成氢键,由此推断,这2种化合物存在活性构象。从3600cm-1附近吸收峰的强度可以看出,HP-3较强,HP-4则相对弱得多,由此表明,邻位桥联屏蔽双酚化合物HP-3存在一定量的活性构象,而邻位桥联屏蔽双酚化合物HP-4则以最低能量构象为主,活性构象较少。
2.3 内旋转能垒对邻位桥联屏蔽双酚构象转变的影响
邻位桥联屏蔽双酚由能量最低构象转变为活性构象,可以更好地发挥抗氧化活性。由于受到分子内原子或基团的相互作用,分子的变形过程会产生一定的内旋转能垒(也称内旋转活化能)[12]。
表6为邻位桥联屏蔽双酚的构象能、活性构象比例估算值和内旋转能垒。图6为邻位桥联屏蔽双酚分子内旋转的过渡态。

图5 3种屏蔽双酚化合物的FT-IR谱Fig.5 FT-IR spectra of three hindered bisphenol compounds

表6 邻位桥联屏蔽双酚的构象能(ΔE)、活性构象比例估算值和内旋转能垒(Ea)Table 6 ΔE,the ratio of active conformation and Eaof hindered ortho-bisphenols

图6 邻位桥联屏蔽双酚分子内旋转的过渡态(TS)Fig.6 The transition-state(TS)of intramolecular rotation of hindered ortho-bisphenols
由表6可知,HP-3活性构象的构象能为11.46kJ/mol,依据Boltzmann分布估算得到的活性构象相对比例约为1%,可见HP-3存在一定量的活性构象。而HP-4活性构象的构象能较高,约为24kJ/mol,活性构象在构象分布中可忽略不计。HP-3的内旋转能垒为72.16kJ/mol,而 HP-4的内旋转能垒则高达129.24kJ/mol。这表明,HP-3的最低能量构象较容易通过分子内旋转变形为活性构象,而HP-4则相对较难。
由图6可知,在分子变形过程中,HP-3的C7—C8键旋转角度很大,而HP-4因受到桥联结构中亚乙基的影响,其C7—C8键旋转的角度较小,C5—C7键旋转的角度较大,从而增加了内旋转难度。这可能是导致HP-3较易内旋转,而HP-4较难内旋转的主要原因。
综合上述分析,HP-3主要通过活性构象来发挥抗氧化作用,其EBD更接近325kJ/mol。而HP-4则很可能主要通过最低能量构象发挥抗氧化作用,其EBD可能处于稍低于379kJ/mol的水平。
因此,在考虑了分子内氢键和内旋转的影响因素时,由EBD反映出的3种屏蔽双酚供氢能力强弱顺序为 HP-3、HP-5、HP-4,这与它们在加氢基础油中抗氧化活性的强弱顺序一致。
3 结 论
(1)由于分子内氢键的影响,邻位桥联屏蔽双酚的抗氧化活性构象是其局部最低能量构象,而不是最低能量构象。
(2)邻位桥联屏蔽双酚的最低能量构象EBD较高,主要是由于2个酚羟基氢原子均参与形成了O—H┅π分子内氢键,受到的束缚作用增强;而局部最低能量构象EBD较低,则主要是因为仅有1个酚羟基的氢原子参与形成了O—H┅O分子内氢键。
(3)内旋转能垒较低,更利于邻位桥联屏蔽双酚由最低能量构象转变为EBD较低的活性构象,这是邻位亚甲基桥联屏蔽双酚抗氧化活性显著高于邻位亚乙基桥联屏蔽双酚的主要原因。
(4)在考虑了分子内氢键和内旋转的影响情况下,屏蔽酚的EBD可以合理表征其在加氢基础油中抗氧化活性,EBD越小,其抗氧化活性越高,与PDSC氧化诱导期数据的规律相一致。为润滑油屏蔽酚抗氧剂的设计和筛选提供了一定理论指导。
[1]WANG Lanfen,SONG Yuguang,ZHANG Xiufeng,et al.An exploratory theoretical elucidation on the peroxyl-radicalscavenging mechanism and structure-activity relationship of nonsteroidal anti-inflammatory drugs [J]. Bioorganic Medicinal Chemistry Letters,2006,16(12):3241-3244.
[2]BALASUNDRAM N,SUNDRAM K, SAMMAN S.Phenolic compounds in plants and agri-industrial byproducts:Antioxidant activity,occurrence,and potential uses[J].Food Chemistry,2006,99(1):191-203.
[3]LUNDBÄCK M,HEDENQVIST M S,MATTOZZI A,et al.Migration of phenolic antioxidants from linear and branched polyethylene [J].Polymer Degradation and Stability,2006,91(7):1571-1580.
[4]RUDNICK L.Lubricant Additives Chemistry and Applications[M].Second edition.New York:Taylor&Francis Group,2009:3-50.
[5]AMORATI R,FUMO M G,MENICHETTI S,et al.Electronic and hydrogen bonding effects on the chainbreaking activity of sulfur-containing phenolic antioxidants[J].The Journal of Organic Chemistry,2006,71(17):6325-6332.
[6]SASON S,AVITAL S.Valence bond diagrams and chemical reactivity [J]. Angewandte Chemie International Edition,1999,38(5):586-625
[7]潘立新,张干兵,曹泽星.羰基镍簇 Ni(CO)n(n=1~4)的结构和Ni-CO键解离性质的密度泛函理论研究[J].高等学校化学学报,2006,27(6):1327-1331.(PAN Lixin, ZHANG Ganbing, CAO Zexing. Density functional calculations on structures and Ni-CO dissociation energies of Ni(CO)n(n=1-4)[J].Chemical Journal of Chinese,2006,27(6):1327-1331.)
[8]TOMIYAMA S,SAKAI S,NIHIYAMA T,et al.Factors influencing the antioxidant activities of phenols by an Ab initio study [J].The Chemical Society of Japan,1993,66(1):299-304.
[9]WRIGHT J S,CARPENTER D J,MCKAY D J,et al.Theoretical calculation of substituent effects on the O—H bond strength of phenolic antioxidants related to vitamin E[J].Journal of the American Chemical Society,1997,119(18):4245-4252.
[10]LUCARINI M,PEDRIELLI P,PEDULLI G F,et al.Bond dissociation energies of O—H bonds in substituted phenols from equilibration studies[J].The Journal of Organic Chemistry,1996,61(26):9259-9263.
[11]AMORATI R,LUCARINI M,MUGNAINI V,et al.Antioxidant activity ofo-bisphenols: The role of intramolecular hydrogen bonding [J].The Journal of Organic Chemistry,2003,68(13):5198-5204.
[12]徐学军,薛英,谢代前,等.Si2Cl6分子的内旋转能垒和振动基频的理论研究[J].化学学报,1999,57(7):680-684.(XU Xuejun,XUE Ying,XIE Daiqian,et al.Computational studies of the structure and vibrational spectra of hexachlorodisilane[J].Acta Chimica Sinica,1999,57(7):680-684.)
