溴酸钾-二甲酚橙体系-顺序注射-催化光度法测定痕量钒
2014-12-01赵诗静李红丽范世华
赵诗静 李红丽 范世华
(东北大学 理学院 分析科学研究中心,沈阳110004)
0 前言
钒是包括人在内的动物体内必需的微量元素,人体中钒的浓度偏低或偏高都会影响人机体的新陈代谢,甚至可能引发某些病变[1]。此外,钒常被用作工业生产中的催化剂或者添加剂,导致钢铁、石油、化工、染料、电子等各类工业废水中钒含量较多,也易造成环境水和土壤污染。准确测定环境水、食品,生物等样品中的痕量钒,具有重要的意义。文献报道的测定钒的方法有分光光度法[2],电感耦合等离子体原子发射光谱法(ICP-AES)[3],电感耦合等离子体质 谱法 (ICP-MS)[4-6],高效液相色谱-质 谱 法(HPLC-MS)[7],石墨炉原子吸收光谱法[8-9],电化学分析法[10]等。上述方法中,或所用仪器较为昂贵,不易普及,或因灵敏度较低,限制了实际应用。
催化动力学分光光度分析方法具有灵敏、简单、快速的特点,是痕量物质分析的重要方法之一,已用于钒的检测[11-13]。但催化光度法一般须采用高温水浴加热,操作不便。包括顺序注射分析在内的流动分析作为一种溶液处理技术,打破了上百年来分析化学反应必须在化学平衡条件下完成的传统,使非平衡条件下的测定成为可能,因而明显提高了分析速度,已用于分光光度分析[14-16]。本文研究了在硫酸介质中,钒(Ⅳ)能明显地催化溴酸钾氧化二甲酚橙指示剂的褪色反应,建立了顺序注射进样条件下催化光度分析测定痕量钒(Ⅳ)的新方法。所建立的分析方法具有灵敏度高、试剂和试样消耗少、分析速度快、操作简便的特点。方法线性范围为0.50~50ng/mL,检出限为0.4ng/mL。将该方法用于环境水中痕量钒(Ⅳ)的测定,分析结果令人满意。
1 实验部分
1.1 仪器与试剂
FIAlab-3000顺序注射分析系统 (FIAlab Instruments,美国):由带三通阀的注射泵、5mL注射器和8通道多位自动选择阀(Valco Instruments,美国)组成;T6新世纪紫外可见分光光度计(北京普析通用仪器有限责任公司)。
钒(Ⅳ)标准储备溶液(100μg/mL):准确称取VOSO4·2H2O 试剂0.039 0g,用二次去离子水溶解于烧杯中,定容至100mL容量瓶中,使用时逐级稀释。
二甲酚橙(XO)指示剂溶液(0.01mol/L):准确称取0.760 6g二甲酚橙,用二次去离子水溶解于烧杯中,然后定容至100mL容量瓶中。使用时逐级稀释。
H2SO4溶液(0.3mol/L),KBrO3溶液(0.3mol/L)。所用试剂均为分析纯,实验用水为二次去离子水。
1.2 实验方法
按照图1所示的实验流路,先吸入一定体积的去离子水作为载流,然后将一定体积的试样溶液、二甲酚橙溶液、溴酸钾溶液三个区带顺序地吸入到储存管道后,改变泵的运行方向将混合后的溶液推至检测器,在440nm处测定反应产物的吸收信号。全部操作过程在计算机控制下按一定程序完成。
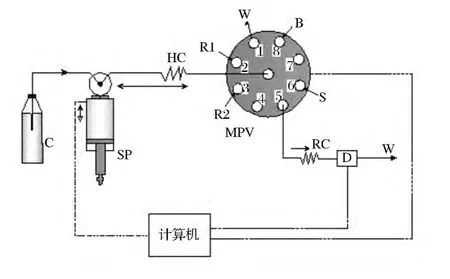
图1 顺序注射光度分析测定钒的流路示意图Figure 1 Schematic diagram of sequential injection spectrophotometry analysis of V(Ⅳ).SP—注射泵;VSP—双位选择阀;MPV—多位阀;HC—储存管路;RC—反应管路;D—检测器;S—样品区带;R1—二甲酚橙(XO)溶液;R2—溴酸钾溶液;B—空白溶液;W—废液
2 结果与讨论
2.1 吸收波长选择
反应产物的吸收光谱见图2。在400~700nm范围内,含有钒(Ⅳ)的催化反应体系和试剂空白的非催化反应体系的最大吸收波长均在440.0nm处,实验最后选择440nm作为检测波长。

图2 吸收光谱曲线Figure 2 Absorption spectra.
2.2 进样顺序
考虑到顺序注射分析系统固有的特点,结合化学反应的具体情况,将试样溶液和硫酸溶液预先混合作为一个区带,二甲酚橙指示剂和溴酸钾溶液(作为氧化剂)分别作为一个区带,经过筛选后采用三区带方式进样。实验观察到,采用“V(Ⅳ)→XO→KBrO3”(进入检测器)的进样方式的吸光度值较大,故在后续的研究中采用“V(Ⅳ)→XO→KBrO3”的进样顺序。
2.3 检测流速
在20~70μL/s的范围内改变检测流速,实验观察到,随着泵速的增加,反应产物的吸光度值迅速增大,达到50μL/s后又开始逐渐降低,这可能是由于显色产物在反应管路中的驻留时间短,难以进行扩散,致使反应进行不完全的缘故(图3)。实验最终选择测定流速为50μL/s。

图3 检测流速对吸光度影响Figure 3 Effect of the flow-rate on absorbance .
2.4 二甲酚橙(XO)区带体积
在50~150μL范围内考察了XO区带体积对吸光度的影响。实验结果如图4所示,溶液的吸光度值随着XO溶液区带体积的增加而增大,当XO区带体积大于100μL时,吸收信号明显降低,这是由于XO区带位于在三区带的中间,进样体积太大,三个试剂区带混合不好,影响试剂充分接触,从而影响吸光度。故实验选择XO溶液的体积为100μL。
2.5 溴酸钾(KBrO3)区带体积
实验结果表明,在175~300μL范围内,随着KBrO3溶液体积增加,吸收信号逐渐增大,当溶液体积为275μL时,吸光度值达到最大,而后继续增加区带体积,吸光度值开始明显降低,最后选择KBrO3溶液进样体积为275μL(图5)。
2.6 试样体积
在150~350μL范围内考察了试样体积对吸收信号的影响。结果表明,随试样体积的增加,吸光度值随之增加,当试样体积增加到300μL时,吸光度值最大。继续增加试样区带体积,吸光度值明显降低(图6)。实验中选择试样区带体积为300μL。

图4 XO进样体积对吸光度的影响Figure 4 Effect of the injection volume of XO reagent on absorbance.

图5 KBrO3进样体积对吸光度的影响Figure 5 Effect of the injection volume of KBrO3 on absorbance.
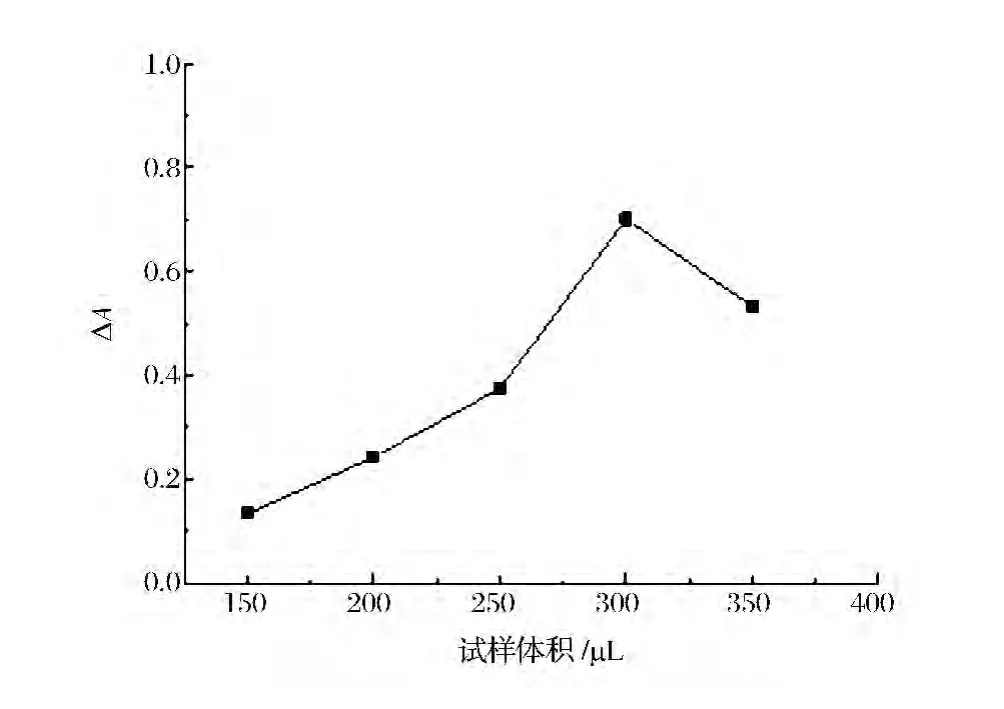
图6 试样区带体积对吸光度的影响Figure 6 Influence of the volume of sample zone on absorbance.
2.7 溴酸钾(KBrO3)浓度
实验考察了KBrO3浓度对吸光度的影响。如图7所示,在0.15~0.30mol/L的范围内,随着KBrO3浓度增加,吸光度值不断增大。鉴于该试剂的溶解度有限,继续增加浓度,试剂配制困难,实验选择KBrO3浓度为0.30mol/L。

图7 KBrO3浓度对吸光度的影响Figure 7 Effect of the concentration of KBrO3on absorbance.
2.8 二甲酚橙(XO)浓度
XO浓度对吸光度的影响如图8所示,随着XO浓度增加,反应产物的吸光度在0.3~0.7mmol/L范围内持续增大,继续增加浓度,尽管吸光度值增大,但由于试剂空白的吸光度值太高,信号不稳,导致测定精度下降,实验选择XO浓度为0.60mmol/L。

图8 XO浓度对吸光度的影响Figure 8 Effect of the concentration of XO on absorbance.
2.9 硫酸(H2SO4)浓度
钒(Ⅳ)对溴酸钾与二甲酚橙的褪色反应的催化过程需要在酸性介质中进行,实验在0.2~0.7mol/L的范围内考察了硫酸溶液浓度对反应产物吸收信号的影响。在实验中观察到,吸光度值随硫酸浓度增大而增加,当硫酸浓度增至0.6mol/L,吸光度值最大,尔后急剧下降(图9)。因此,实验选择硫酸的浓度为0.60mol/L。

图9 H2SO4浓度对吸光度的影响Figure 9 Effect of the concentration of H2SO4 on absorbance.
2.1 0 共存组分的干扰
在相对误差±5%范围内,试样溶液中可能的共存物质对50ng/mL钒(Ⅳ)测定的影响列于表1。其中,V(Ⅴ),NO-2,Fe2+,Pb2+等离子干扰较大,测定时需要预先除去。
2.1 1 方法的分析性能
在选定实验参数下,钒(Ⅳ)浓度在0.5~50ng/mL范围内与吸光度呈良好的对数线性响应关系。线性回归方程为lgA0/A=0.008×lgC+0.013 7,相关系数R=0.999 0。方法的相对标准偏差(RSD)为1.1%(n=11)。以空白溶液吸光度的3倍标准偏差与标准曲线斜率的比值作为方法的检出限(3σ),计算得出钒(Ⅳ)的检出限为0.4ng/mL。
2.1 2 实际样品的测定
2.1 2.1 试样前处理
取50mL过滤后的水样,向该水样中加入1mL的EDTA(50g/L)和2mL的NaF(50g/L)掩蔽试剂,放置反应1h。然后向上述处理后的溶液中加入一定量的银微粒,加入一定量 HCl(0.5mol/L),加热到60℃反应0.5h。过滤除去银微粒,调节pH值至中性,最后将溶液定容至25mL。用同样方法对空白进行处理。
2.1 2.2 实际样品测定
取处理后的试样溶液5mL置于10mL容量瓶中,按实验方法测定,同时进行加标回收实验,实验结果列于表2。
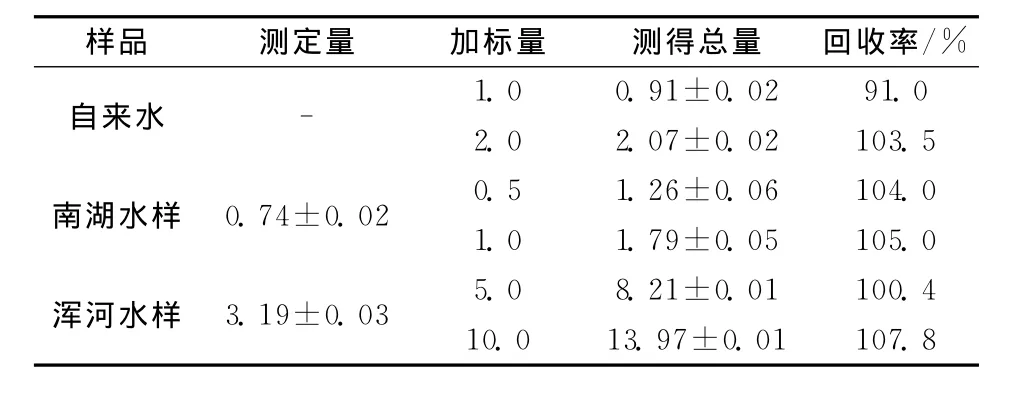
表2 环境水样中钒的测定结果Table 2 Analytical results for vanadium in environmental water samples /(ng·mL-1)
3 结语
在常温下研究了硫酸介质中钒(Ⅳ)对溴酸钾-二甲酚橙氧化反应体系的催化作用,建立了顺序注射催化光度分析测定环境水中痕量钒的新方法;该法在常温下测定,灵敏度高,分析速度快,试剂消耗少,每小时可分析30个水样;在440nm处钒(Ⅴ)也可发生催化反应,但灵敏度不如钒(Ⅳ),钒(Ⅳ)和钒(Ⅴ)的同时测定正在研究中。
[1]吕选忠,于宙,王广仪,编著 .元素生物学[M].合肥:中国科学技术大学出版社,2011:388-400.
[2]尹继先,朱霞萍,梁庆勋,等 .微波消解-分光光度法测定钒钛磁铁矿中的钒[J].岩矿测试,2010,29(6):719-722.
[3]成勇,袁金红,彭慧仙,等 .微波消解-电感耦合等离子体原子发射光谱法测定钒钛磁铁矿中锆铌钒铬[J].冶金分析,2013,33(12):43-46.
[4]Chirinos J,Oropeza D,González J,et al.Determination of vanadium/nickel proportionality in the asphaltene frac-tion of crude oil using thin-layer chromatography with femtosecond laser ablation-inductively coupled plasmamass spectrometry[J].Energy&Fuels,2013,27(5):2431-2436.
[5]Kilibarda N,Afton S E,Harrington J M,et al.Rapid speciation and determination of vanadium compounds using ion-pair reversed-phase ultra-high-performance liquid chromatography inductively coupled plasma-sector field mass spectrometry[J].Journal of Chromatography A,2013,1304:121-126.
[6]成勇 .电感耦合等离子体质谱法(ICP-MS)测定油品中铁、铜、铅、锡、砷、银、铬、镍、钒[J].中国无机分析化学,2011,1(4):64-67.
[7]Nischwitz V,Davies J T,Marshall D,et al.Speciation studies of vanadium in human liver(HepG2)cells after in vitro exposure to bis(maltolato)oxovanadium(Ⅳ)using HPLC online with elemental and molecular mass spectrometry[J].Metallomics,2013,5(12):1685-1697.
[8]Dobrowolski R,Adamczyk A,Otto M.Determination of vanadium in soils and sediments by the slurry sampling graphite furnace atomic absorption spectrometry using permanent modifiers[J].Talanta,2013,113:19-25.
[9]Wadhwa S K,Tuzen M,Kazi T G,et al.Graphite furnace atomic absorption spectrometric detection ofvanadium in water and food samples after solid phase extraction on multiwalled carbon nanotubes[J].Talanta,2013,116:205-209.
[10]梁庆勋,朱霞萍,陈文,等.极谱法测定钒钛磁铁矿中钒[J].冶金分析,2011,31(6):31-33.
[11]陈亚红,田丰收,黄凤羽 .酶催化分光光度法测定环境水中痕量钒[J].冶金分析,2014,34(1):59-62.
[12]樊雪梅 .催化动力学光度法测定钒尾矿中痕量钒(Ⅴ)[J].分析科学学报,2013,29(3):419-421.
[13]王艳丽 .催化动力学分析法测定超痕量钒的研究[J].分析测试技术与仪器,2010,16(1):42-47.
[14]孙菲菲,范世华 .在线稀释分光光度法测定人体尿液中的肌酸酐[J].分析试验室,2013,32(11):50-53.
[15]王夕云,陈志霞,范世华 .顺序注射催化光度分析法测定环境水样中痕量钼[J].中国无机分析化学,2011,1(3):27-31.
[16]李潇 .顺序注射-分光光度测定联用技术在元素形态分析中的应用[D].沈阳:东北大学,2010.
