Illumina高通量测序技术分析早产儿出生后肠道菌群变化的初步研究
2014-11-21方剑火郎继东
陈 娜 杨 毅 张 澜 方剑火 郎继东 曹 云 田 埂,3
肠道菌群履行着重要的功能,如屏障功能、代谢反应、营养作用、宿主固有免疫和适应性免疫应答成熟等,与人类的健康密切相关[1]。已有研究显示早产儿肠道菌群的建立与晚发型败血症(LOS)和坏死性小肠结肠炎(NEC)有关[2]。尽管NEC的确切病因尚不清楚,但普遍认为细菌的异常定植是重要原因之一[3],而许多LOS由肠源性细菌易位引起[4],并且肠道屏障的改变也会促成LOS和NEC的发生。早产儿由于各脏器发育不成熟,生后多处于NICU中,受到应用抗生素、缺乏母乳喂养以及医疗操作(如肠外营养、侵入性操作)等多方面的影响,导致其肠道菌群定植的模式及数量发生异常改变[5]。
研究表明能进行培养的细菌仅占肠道菌群的1%~10%[6],传统培养法不能充分反映菌群的多样性。变性梯度凝胶电泳(DGGE)[7,8]、实时荧光定量 PCR[9]等非培养技术被用于肠道菌群的研究,但只能检测出一定丰度以上的细菌或特定微生物。近年来,高通量测序技术具备测序快、准确度高等特点,已成为研究肠道微生物的主要研究方法[10,11],国外有报道采用该方法探讨了早产儿肠道菌群与败血症的关系[10,12],国内尚未见相关研究报道。
1 方法
1.1 研究设计 本研究采集早产儿生后不同时点的粪便样本,采用Illumina高通量测序技术分析肠道菌群物种丰度、多样性和构成的变化趋势,探讨住院期间发生全身炎症反应综合征(SIRS)或败血症或NEC早产儿肠道菌群定植模式。本文描述性报道预试验结果。
1.2 纳入和排除标准 ①2014年2~3月复旦大学附属儿科医院NICU住院的早产儿,胎龄≤34周或出生体重≤1 750 g;②排除住院时间<7d患儿;③排除合并严重先天性心脏病、严重消化道畸形需手术的早产儿,排除唐氏综合征、遗传代谢病和重度窒息的早产儿。
1.3 SIRS和败血症诊断标准[13,14]SIRS定义为:①体温>38.5℃或<36℃;②心动过速(平均心率>正常同年龄标准的2 s),心动过缓(平均心率<正常同年龄标准的P10);③呼吸急促(平均呼吸频率>正常同年龄标准的2 s)或急性期机械通气;④WBC升高或降低或CRP>10mg·L-1。败血症定义为SIRS的基础上血培养阳性。
1.4 标本采集 于生后第1天采集胎粪,之后计划于每周龄时或评估败血症时采集粪便样本,直至出院或生后8周。采用无菌通便的方法采集粪便,用无菌棉签挑取新鲜粪便放入无菌冻存管后立即速冻,送至实验室提取DNA,余粪便样本-80℃保存。
1.5 DNA提取 采用Powerfecal DNA Isolation Kit(MoBio,美国)试剂盒提取100mg粪便样本中微生物的总DNA,具体步骤按说明书操作。所提取的DNA于-20℃保存备用。1.6 16S rRNA-V3区的PCR扩增及测序 用提取的总DNA作为模板,16S PCR引物由测序接头引物、Index和V3区引物3部分组成。Index为6 bp核苷酸随机组成的序列,以标记PCR产物的来源。PCR扩增体系采用25μL:2 ×Master Mix(NEB,美国)12.5μL,引物各 1.5μL,模板2.5μL,灭菌双蒸水7μL(如模板DNA浓度较低则不加灭菌双蒸水,加等体积的模板)。扩增条件:98℃预变性30 s,98 ℃ 10 s,50℃ 30 s,72℃ 30 s,20 个循环,最后72℃延伸7 min,4℃保存。反应结束后将全部反应产物用1.5%的琼脂糖凝胶电泳(溴化乙锭染色)检测扩增片段大小,采用QIAquick Gel Extraction Kit(QIAGEN,德国)对目的条带胶回收纯化。
将纯化后的16S rRNA-V3区的PCR产物送至清华大学医学院进行测序,测序平台为Illumina Miseq。
1.7 生物信息学分析 将测序数据上传至MG-RAST服务器端 (http://metagenomics.anl.gov/)[15],应用 MG-RAST V3.3.6中的rRNAdetection pipeline进行数据分析,原始数据经过预处理去除低质量的序列后,按照rRNA发现、聚类和鉴定的步骤进行分析。计算在97%的相似水平上每个样本的操作分类单元(OTU)数量,特定的分类单元代表某特定物种,基于样品测序产生的OTU的结果绘制稀疏曲线。计算单个样本的Alpha多样性(Shannon指数),指数越大表示该样本中的物种越丰富。在门和科两个分类水平上统计样本的物种丰度,并行聚类分析。
1.8 临床资料截取 从病史中截取母亲孕期的信息(胎膜早破、感染,妊娠高血压、抗生素使用等),喂养方式(母乳喂养或配方奶喂养),达足量喂养日龄,抗生素暴露,使用益生菌情况,感染(SIRS和败血症)和NEC发生情况。
2 结果
2.1 一般情况 3例早产儿共采集了生后1、7、14和21d的12份粪便样本,其中3例生后1d、例1生后7d和例2生后14d粪便样本因PCR扩增失败,进入测序分析的粪便样本为7份。3例早产儿平均胎龄为(31.3±0.8)周,平均出生体重为(1 540±144)g。例1母亲有胎膜早破19h伴发热病史,产前有抗生素暴露史。3例早产儿生后均使用早产儿配方奶喂养,但达足量喂养的日龄有差别。3例早产儿生后早期均使用抗生素,但使用时间均<7d。例2在生后26d诊断SIRS,并应用抗生素治疗,但分析的粪便样本在感染前采集(表1)。
2.2 物种丰度及多样性分析 7份粪便样本的序列数分别为1 505 736(例 1生后 14d)、1 695 090(例 1生后21d)、241 694(例2 生后7d)、1 186 332(例2 生后21d)、1 193 486(例3生后7d)、1 082 270(例3生后14d)和671 600(例3生后21d)。
样本稀疏曲线(图1)显示,在<84 750条序列时,OTU数量随序列数增加而迅速增加;在~339 000条序列时,OTU数目增加缓慢;之后则趋于平台期。
各样本的OTU数量:例1生后14和21d分别为533和608,例2生后7和21d分别为381和576,例3生后7、14和21d分别为409、571和524;各样本的Shannon指数:例1生后14和21d分别为2.61和7.15,例2生后7和21d分别为2.00和4.28,例3生后7、14和21d分别为7.69、7.04和8.44;均随日龄增加呈升高趋势。例2的2个时点样本的Shannon指数均低于相同时点的例1和(或)例3样本。

表1 3例早产儿的基本临床特征Tab 1 Basic clinical characteristics of 3 infants
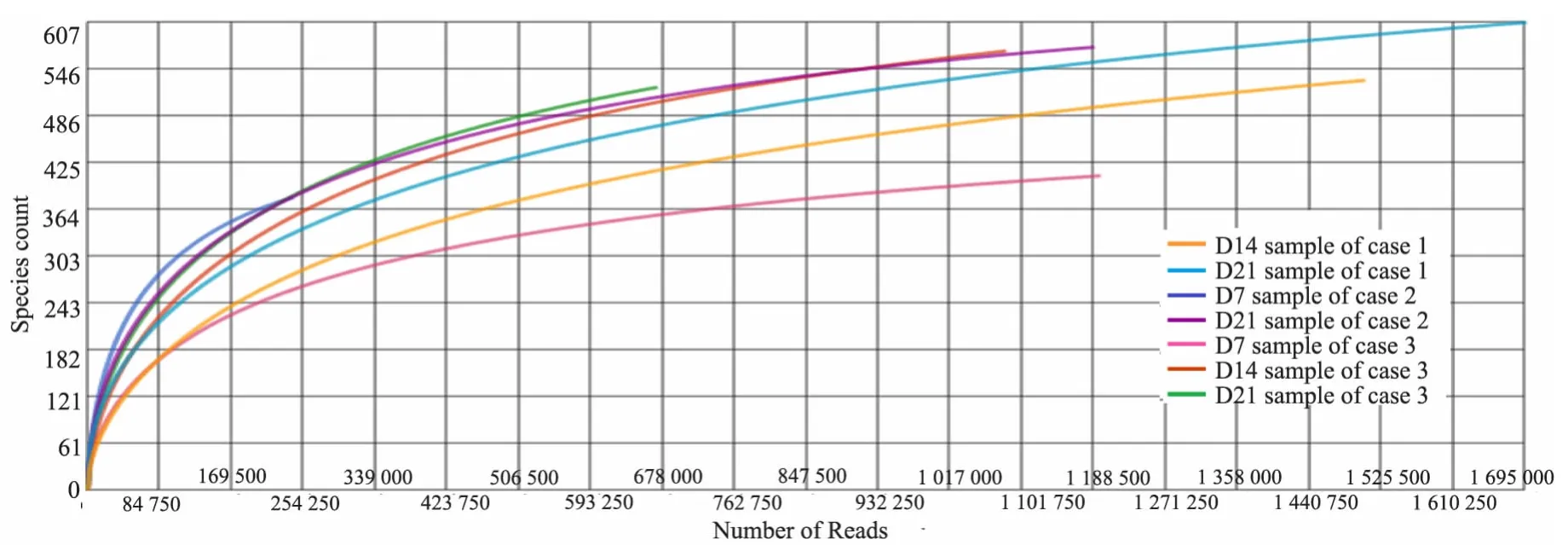
图1 7份粪便样本的稀疏曲线Fig 1 Rarefaction analysis of 7 samples
2.3 门水平下早产儿肠道菌群结构 例1肠道菌群的组成不同于例2和3,以变形菌门占优势(66.94%vs 0.27%vs 0.14%),其次为厚壁菌门(32.63%)。例2和3肠道菌群的组成相似,均以厚壁菌门占优势(分别占75.22%和83.51%),其次为放线菌门(分别占24.51%和16.35%)。
7份样本共检测到18个菌门,每个样本检测到10~13个菌门,其中放线菌门、拟杆菌门、蓝藻菌门、异常球菌-栖热菌门、厚壁菌门、梭杆菌门、变形菌门及柔壁菌门为共有菌门。表2显示,7份样本99.9%以上的序列主要是放线菌门、拟杆菌门、厚壁菌门和变形菌门,但这4种优势菌门在各样本中所占比例存在差异。变形菌门在例1生后14d和21d样本中分别占97.52%和49.11%,放线菌门在例2生后7d样本中占99.46%,余4份样本以厚壁菌门丰度最高(分别占93.85%、77.42%、84.67%和94.21%)。
2.4 科水平下早产儿肠道菌群结构 7份样本共检测到172个科,每个样本检测到96~122个科,其中63科为7份样本共有。对丰度≥1%的科进行了统计,<1%的计入其他。表3显示,7份样本中相对丰度较高的科共检测到10个,除例1生后14d标本以肠杆菌科为绝对优势菌(96.46%),例2生后7d样本以棒状杆菌科为绝对优势菌(97.90%)外,其余样品菌群的分布相对均匀。值得注意的是,例2生后21d样本中葡萄球菌科所占比例较高(27.16%),分类到种水平主要为金黄色葡萄球菌和里昂葡萄球菌(分别占葡萄球菌科的71.5%和18.3%)。
2.5 早产儿肠道菌群结构聚类 由于鉴定出的菌门较少,而科水平较多,不宜行聚类分析,故在纲水平对7份样本进行聚类分析,并构建系统树状图(图2),例1的2份、例2的2份和例3的3份样本分别聚为一簇;7份样本共鉴定出39个菌纲,其余11个为未分类序列和未分配序列,共聚为5簇;丰度较高的菌群主要有放线菌纲,芽孢杆菌纲,梭菌纲,Negativicutes,α、β、γ、δ变形菌纲。
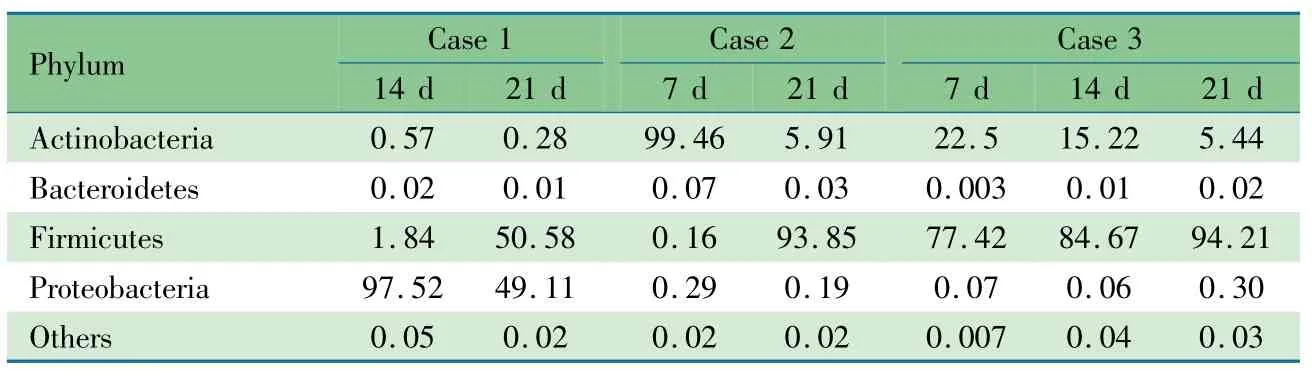
表2 3例早产儿不同时点粪便样本在门水平的肠道菌群组成(%)Tab 2 Gut bacterial composition at phylum level per sample(%)
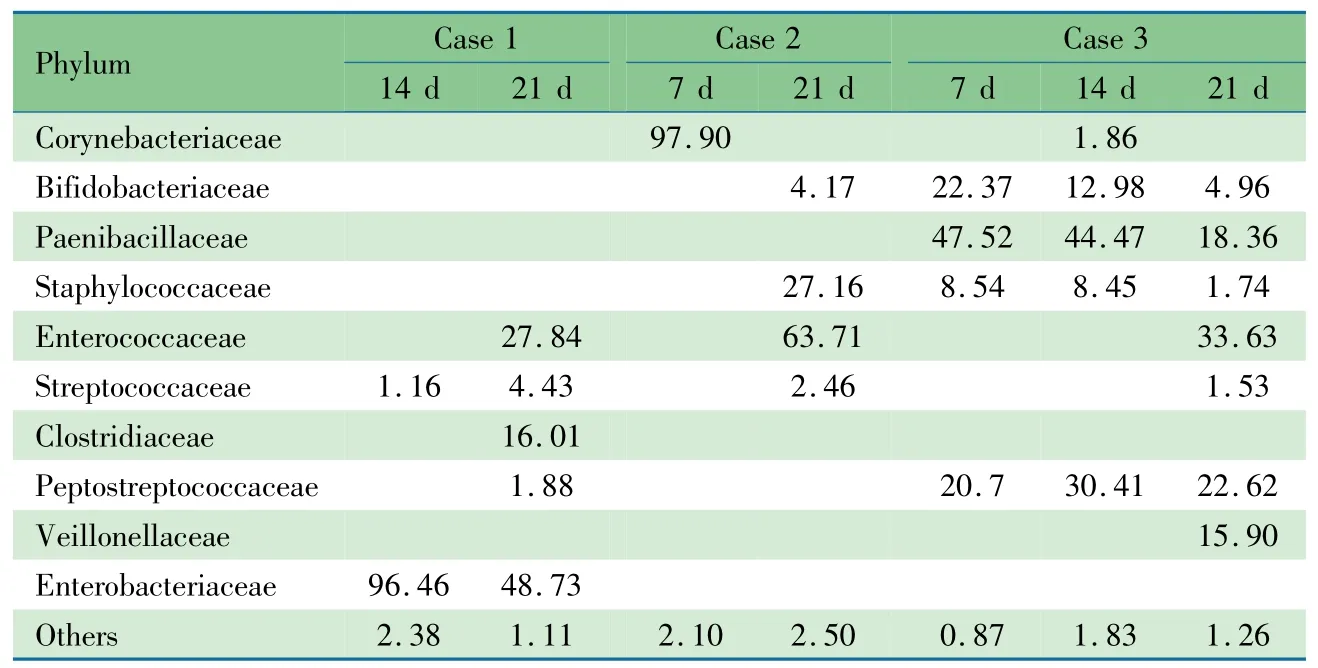
表3 3例早产儿不同时点粪便样本在科水平的肠道菌群组成(%)Tab 3 Gut bacterial composition at family level per sample(%)

图2 3例早产儿不同时点粪便样本在纲水平的聚类图Fig 2 Heatmap at class level per sample atdifferent time points
3 讨论
与传统的培养方法及16S rDNA为基础的分子生物学方法不同,高通量测序技术基于16S rDNA的微生物群落分析中的要点,在于产生测序覆盖深度极深的测序数据,并通过比对或聚类的分析方法,对数据来源的微生物物种进行分析,并估计微生物群落的物种构成[16]。Illumina高通量测序技术是目前用于肠道微生物研究最前沿的检测技术[17],可以发现低丰度细菌或未知细菌,进而全面、准确地获得菌群的信息。
新生儿肠道菌群的建立是一个缓慢渐进的动态演变过程。通常认为胎儿肠道是无菌的,但有证据表明宫内环境有细菌存在,提示这些细菌可能影响新生儿出生前的微生物群[18,19]。早产儿胎粪中的细菌可能是宫内来源的,反映了宫内感染、继发于早产胎膜早破的定植或母亲抗生素应用等宫内暴露[10]。本研究3例早产儿胎粪样本的细菌总DNA浓度非常低,16S rRNA-V3区 PCR扩增失败,因此无法观察到胎粪细菌的情况,当然也不能排除DNA提取的问题。
新生儿肠道菌群的建立和演变受多种因素的影响,如地理环境、胎龄、分娩方式、喂养方式、卫生状况及抗生素应用等。与足月儿相比,早产儿肠道正常菌群的定植明显延迟,达优势化时间也延迟[20],且肠道菌群多样性也较低。有研究表明某些微生物定植肠道存在一个胎龄临界值,对于双歧杆菌这一临界值约为33周[21]。不同喂养方式的新生儿肠道菌群定植也存在差异。母乳喂养儿粪便中双歧杆菌的量明显高于人工喂养儿[22]。本文3例早产儿均采用配方奶喂养,但例3粪便样本中双歧杆菌的相对丰度要高于例1和2,回顾临床资料发现例3胎龄最大,开奶时间也最早,且喂养和住院过程较顺利,促进了消化道发育成熟,有利于早期形成完善的肠道微环境,使双歧杆菌得以定植和繁殖。
长期大量应用抗生素对肠道正常菌群可造成损害,降低了正常菌群的定植抗力,有利于潜在致病菌的生长,引起肠源性感染。应用抗生素的新生儿,其粪便中肠杆菌、肠球菌的比例明显增加,双歧杆菌的比例下降,有些差异在抗生素治疗结束后1个月仍然存在[23]。低出生体重儿早期应用抗生素可显著降低肠道菌群的多样性[10]和粪便微生物的总数[24]。本文发现例1生后14d粪便样本中肠杆菌科占绝对优势(主要是克雷伯菌属),双歧杆菌科比例很小(<1%),肠道菌群的多样性也较低,回顾资料发现,该患儿母亲产前因胎膜早破伴发热应用了抗生素治疗,且患儿于生后24h内应用了两联广谱抗生素治疗6d,可能与上述结果有关。尽管许多外部因素会对早产儿肠道菌群产生影响,但本文通过聚类分析发现同一个体不同时点肠道微生物构成相似性较高,提示早产儿生后早期肠道菌群的构成情况可能受遗传因素或母亲因素的影响要高于外部因素对其产生的影响。
本研究分析的3例早产儿住院过程中未发生NEC,例2在住院过程中发生了SIRS,其生后早期肠道菌群的多样性一直处于较低水平,在感染发生前5d(生后21d)采集的粪便样本发现了比例较高的葡萄球菌科(27.16%),且主要为致病性的金黄色葡萄球菌,其条件致病菌肠球菌科所占的比例也显著高于例1和3粪便样本(63.71%vs 27.84%vs 33.63%)。Madan等[10]研究也发现发展为败血症的早产儿其生后肠道微生物多样性较低,且以葡萄球菌占优势。葡萄球菌为共生菌,但当其大量存在时也会成为易位细菌,从而成为NICU内葡萄球菌败血症常见风险的影响因素[25]。由于本文分析的病例数较少,初步探讨了早产儿肠道菌群改变与败血症发生的关系,还需要更多样本来验证这些结果。
总之,NICU内早产儿肠道菌群的定植和演变受内部因素和外部因素的影响发生了许多改变。经验性、长期应用广谱抗生素会显著降低肠道菌群的多样性,影响正常菌群的定植。发展为败血症的早产儿生后肠道微生物多样性较低,以致病菌占优势的肠道微生物区可能与败血症的发生相关。
[1]Matamoros S, Gras-Leguen C, Le Vacon F, et al.Development of intestinal microbiota in infants and its impact on health.Trends Microbiol, 2013,21(4):167-173
[2]Berrington JE, Hearn RI, Bythell M, et al.Deaths in preterm infants:changing pathology over 2 decades.J Pediatr, 2012,160(1):49-53
[3]Grishin A, Papillon S, Bell B, et al.The role of the intestinal microbiota in the pathogenesis of necrotizing enterocolitis.Semin Pediatr Surg, 2013,22(2):69-75
[4]Vergnano S, Menson E, Kennea N, et al.Neonatal infections in England:the NeonIN surveillance network.Arch Dis Child Fetal Neonatal Ed, 2011,96(1):9-14
[5]Xu YZ(徐艳珍), Yu JL,Ai Q, et al.The variation of Escherichia coli,Klebsiella pneumoniae and Enterococcus faecalis in preterm infants with feeding intolerance.Chinese Journal of Microecology(中国微生态学杂志), 2012,24(8):727-732
[6]Haraszthy VI, Zambon JJ, Sreenivasan PK, et al.Identification of oral bacterial species associated with halitosis.J Am Dent Assoc, 2007,138(8):1113-1120
[7]Stewart CJ, Marrs EC, Magorrian S, et al.The preterm gut microbiota:changes associated with necrotizing enterocolitis and infection.Acta Paediatr, 2012,101(11):1121-1127
[8]Smith B, Bode S, Skov TH, et al.Investigation of the early intestinal microflora in premature infants with/without necrotizing enterocolitis using two different methods.Pediatr Res, 2012,71(1):115-120
[9]Lei Y(雷毅),Wang KH,Gong KM,et al.Real-time fluorescence quantitative PCR analysis of intestinal flora in patients with acquired immune deficiency syndrome.The Journal of Practical Medicine(实用医学杂志), 2012,28(1):69-71
[10]Madan JC, Salari RC, Saxena D, et al.Gut microbial colonisation in premature neonates predicts neonatal sepsis.Arch Dis Child Fetal Neonatal Ed, 2012,97(6):456-462
[11]Li XM(李晓敏), Yang LJ, Huo GC.Illumina technology research the difference of intestinal flora in infants with different feed fashion.Food Science and Technology(食品科技),2012,(9):319-324
[12]Mai V,Torrazza RM,Ukhanova M,et al.Distortions in development of intestinal microbiota associated with late onset sepsis in preterm infants.PLoS One, 2013,8(1):e52876
[13]Goldstein B, Giroir B, Randolph A.International pediatric sepsis consensus conference:definitions for sepsis and organ dysfunction in pediatrics.Pediatr Crit Care Med, 2005,6(1):2-8
[14]Wynn JL, Wong HR.Pathophysiology and Treatment of Septic Shock in Neonates.Clin Perinatol, 2010,37(2):439-479
[15]Meyer F, Paarmann D, D'Souza M, et al.The metagenomics RAST server-a public resource for the automatic phylogenetic and functional analysis of metagenomes.BMC Bioinformatics,2008,9:386
[16]Liu LY(刘莉扬), Cui HF, Tian G.Application of highthroughput sequencing technology in matagenome.Chin Med Biotechnol(中国医药生物技术), 2013,8(3):196-200
[17]Qin N(秦楠), Li DF, Yang RF.Next-generation sequencing technologies and the application in microbiology.Acta Microbiologica Sinica(微生物学报), 2011,51(4):445-457
[18]Penders J, Thijs C, Vink C, et al.Factors influencing the composition of the intestinal microbiota in early infancy.Pediatrics, 2006,118(2):511-521
[19]Jimenez E, Marin ML, Martin R, et al.Is meconium from healthy newborns actually sterile?Res Microbiol, 2008,159(3):187-193
[20]Zhang L(张琳), Liang QH.Research status of intestinal microecology in neonates.Chinese Journal of Practical Pediatrics(中国实用儿科杂志), 2000,15(12):761-762
[21]Butel MJ, Suau A, Campeotto F, et al.Conditions of bifidobacterial colonization in preterm infants:a prospective analysis.J Pediatr Gastroenterol Nutr, 2007,44(5):577-582
[22]Bezirtzoglou E, Tsiotsias A, Welling GW.Microbiota profile in feces of breast-and formula-fed newborns by using fluorescence in situ hybridization(FISH).Anaerobe, 2011,17(6):478-482
[23]Tanaka S, Kobayashi T, Songjinda P, et al.Influence of antibiotic exposure in the early postnatal period on the development of intestinal microbiota.FEMS Immunol Med Microbiol, 2009,56(1):80-87
[24]Westerbeek EA, van den Berg A, Lafeber HN, et al.The intestinal bacterial colonisation in preterm infants:a review of the literature.Clin Nutr, 2006,25(3):361-368
[25]Lindberg E, Adlerberth I, Matricardi P, et al.Effect of lifestyle factors on Staphylococcus aureus gut colonization in Swedish and Italian infants.Clin Microbiol Infect, 2011,17(8):1209-1215
