含二甲亚枫配体锇配合物的溶剂化显色效应
2014-11-09张建坡郝希云孙秀云
张建坡,金 丽,李 岩,郝希云,孙秀云
(吉林化工学院化学与制药工程学院,吉林吉林132022)
当今过渡金属配合物新材料的合成和器件开发引起科学家的持续关注.具有饱和配位的d6过渡金属配合物,由于可以实现单重态到三重态的隙间穿越导致其具有非常高的磷光量子效率,被迅速应用到生产生活的各个领域[1].传统的研究多以Ir(III)和Pt(II)配合物为主,直到最近几年,人们发现锇配合物独特的结构优势,导致其较低的辐射寿命和更高的磷光量子效率,可以超越传统的磷光材料在OLED上加以应用,锇(II)配合物才逐渐被人们关注[2].
最近,一系列顺式氰基Os(II)配合物[(DMSO)2(CN)2Os(N^N)][N^N=bpy or phbpy;bpy=2,2'-吡啶;phbpy=4,4-双苯基-2,2'-吡啶]被支等人合成[3].实验研究表明该类配合物具有卓越的溶剂化显色效应,优良的吸收和发射性能,以及较高的磷光量子效率.溶剂化显色效应是此类材料的一个突出特点,材料的结构与其性能息息相关.以前也曾有类似的报道,[Ru(bpy)(CN)4]2-和[Fe(diimine)2(CN)2][4-5]配合物,由于氰基配体上存在孤对电子,此孤对电子与溶剂分子相互作用会产生分子间的氢键作用,导致电子密度偏离金属原子,使其在不同溶剂极性下其吸收和发射变得非常敏感.当前,量子化学方法已发展的相当完善和先进,理论计算的发展为新材料的设计和人为调控材料性能开启了新思路.计算化学家不再满足于气相环境下的计算.气相预测虽然在许多方面是很准确的,但是描述分子在溶液中的特征时不够严格[6].因此,本研究计划选取气态(1)和三个有代表性的溶剂二氯甲烷(2)、甲醇(3)和水(4)对[Os(DMSO)2(CN)2(N^N)](N^N=2,2'-吡啶)配合物的结构和光谱性质进行系统的理论研究,通过对不同溶剂下配合物的几何结构、吸收以及发射光谱的深入研究,揭示溶剂极性与配合物结构和光谱性质之间的内在联系.
1 计算方法
配合物在不同溶剂下的基态和激发态结构均采用Cs对称性.采用DFT方法对配合物的基态及激发态的几何结构进行全优化.使用HF/DFT的混合泛函PBE0[7]泛函优化基态几何结构,用非限制性PBE0泛函优化激发态几何结构.以基态以及激发态的优化几何结构为基础,应用TD-DFT方法结合PCM(polarized continuum model)模型得到了配合物在不同溶剂下的分子轨道成分、电荷布居、吸收以及发射光谱.在计算中,对Os原子采用了由Hay和Wadt[8]提出的16价电子准相对论赝势模型,采用LanL2DZ基组,为了更好的描述金属和配体之间的相互作用,对Os原子还加入一个f型极化函数(α=0.886),对于其它原子使用6-31G*基组.以上计算通过执行Gaussian03程序包[9]在IBMX3400M2服务器上完成.
2 结果与讨论
2.1 溶剂极性与分子几何
用PBE0泛函优化配合物[Os(DMSO)2(CN)2(N^N)]的基态结构展示在图1中.在CH2Cl2溶液中Os-S1、Os-C1和Os-N3键长与其实验值分别相差了 0.047、0.011 和 0.003Å[6],键角S1-Os-S2、S1-Os-N3和S1-Os-C1与实验值的差距也都在3.0°之内.以上数据均说明计算结果的准确可靠.在极性较大的溶剂(H2O和CH3OH)中Os-S1、Os-C1键较长,Os-N3键较短,这表明在极性溶剂的作用下使DMSO和CN基配体的电子云密度下降,N^N配体的电子云密度上升,从而增强了金属与N^N配体的相互作用,削弱了金属与DMSO和CN基配体的相互作用.另外,在所有溶剂中键角 S1-Os-S2接近于180°,S1-Os-C1和 S1-Os-N3接近于90°,表明整个分子处于正八面体的稳定结构.以上研究表明:溶剂分子只对配合物的电子云分布有较小影响,对分子的空间主体几何结构影响不大.

图1 配合物[Os(DMSO)2(CN)2(N^N)]的结构简图及原子编号
在激发态下,配合物的键长表现出不规律的变化,极性大的溶剂(H2O和CH3OH)Os-S1键缩短,而Os-N3键增长,这种变化趋势与在气态和二氯甲烷中正好相反.从键角的变化趋势可以看出,极性小的溶剂(CH2Cl2和gas)使分子在激发态下更接近于正八面体结构,极性大的溶剂使分子激发态结构发生扭曲,略微偏离正八面体结构.
2.2 溶剂极性与吸收光谱
在基态几何基础上,利用TD-DFT方法计算了配合物在溶剂1-4中紫外-可见吸收光谱,图2给出了模拟的Gaussian型吸收曲线,将配合物在溶剂1-4中有代表性的光谱,以及它们的振荡强度、CI系数、跃迁性质和所对应实验值列于表1.表2列出了配合物在H2O中的电子吸收跃迁涉及到的部分分子轨道成分.

图2 高斯曲线模拟配合物在gas(1)、CH2 Cl2(2)、CH3 OH(3)和H2 O(4)溶剂中的吸收光谱
从图2、表1和2可以看出,配合物在1-4溶剂中都有三个明显的吸收带,它们的最低能吸收由于振荡强度比较小,在模拟的吸收曲线中观察不到.来自于表1,四个配合物最低能吸收分别出现在519(1)、501(2)、494(3)和485 nm(4).配合物在H2O(4)溶剂下485 nm的吸收来自于分子轨道93→95的激发,分子轨道93由 d(OS)、π(CN)和π(N^N)轨道成分构成;而95是最低空轨道(LUMO),主要由π*(N^N)轨道占据.因此,此跃迁是属于[d(OS)+π(CN)+π(N^N)→π*(N^N)]的金属到配体的电荷转移(MLCT)跃迁,并混有少量的配体到配体的电荷转移(LLCT)跃迁.与在4溶剂中类似,在1-3溶剂中的最低能吸收也是来自于分子轨道93→95的激发,并且它们具有相同的跃迁性质.

表1 配合物在不同溶剂中的紫外可见吸收光谱及其实验值
比较在1-4溶剂中的最低能吸收可以看出,随着溶剂极性的逐渐增大(1<2<3<4),最低能吸收发生明显的蓝移,这与它们逐渐增大的能级间隙相一致.来自于轨道能级分析,配合物在1-4溶剂中分子轨道93→95的能级间隙分别为3.20(1)、3.28(2)、3.32(3)和3.36 eV(4),随着溶剂极性的逐渐增大,能级间隙逐渐增大,该跃迁必然发生蓝移,通过图2可以直观的看到.

表2 配合物在H2 O(4)中的电子吸收跃迁涉及到的部分分子轨道成分
配合物在1-4溶剂中的较强吸收带分别在451(1)、441(2)、436(3)和 430 nm(4),其振子强度分别为 0.074 1、0.083 2、0.083 9 和 0.086 7 类似于上面的分析,配合物在该处跃迁也属于MLCT/LLCT跃迁.比较在1-4溶剂中的强吸收可以看出,随着溶剂极性的逐渐增大,强吸收峰也有较小的蓝移.
配合物在1-4溶剂中的高能吸收带代表性吸收分别在268(1)、269(2)、271(3)和272 nm(4),它们具有最大的振子强度,是实验上最容易观察到的.通过轨道占据分析可知,该跃迁主要发生在DMSO和N^N配体上,具有配体到配体和发生在配体内部的混合电荷转移(LLCT/ILCT)跃迁性质.比较1-4的高能吸收可以看出,溶剂极性对该处吸收的影响最小(<5 nm),且随着溶剂极性增加有略微的红移趋势.比较以上三个吸收带可以看出溶剂的极性往往对最低能吸收的影响更大,对高能吸收基本没有影响.
2.3 溶剂极性与发射光谱
表3还列出了配合物在1-4溶剂中的磷光发射光谱数据.
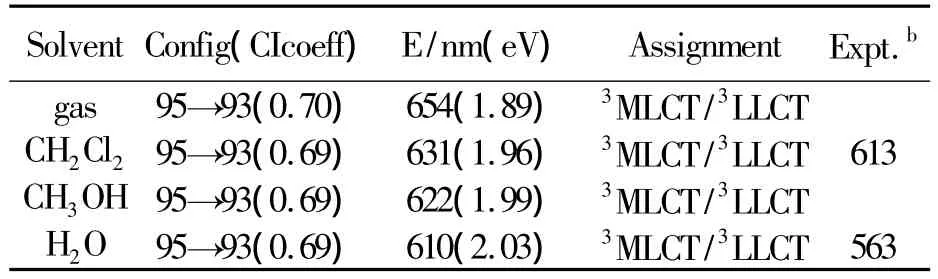
表3 配合物在1-4溶剂中的发射光谱及其实验值
由表3可知,配合物在1-4溶剂中最低能发射分别在654(1)、631(2)、622(3)和 610 nm(4).配合物在4溶剂中610 nm的发射来自于分子轨道95→93的激发,类似上面的轨道占据分析可知,分子轨道93由d(OS)、π(CN)和π(N^N)轨道成分构成,而分子轨道95为最低空轨道(LUMO),主要为π*(N^N)型轨道,因此这个跃迁被指认为3MLCT/3LLCT特征,这与其最低能吸收的跃迁性质相一致.并且配合物在1-3溶剂中的最低能发射也具有相似的3MLCT/3LLCT特征.
比较配合物在1-4溶剂中的最低能吸收和发射,配合物在四个溶剂中有相似的轨道成分和跃迁性质,它们的最低能吸收和发射的能量差分别为 0.50(1)、0.51(2)、0.52(3)和 0.48 eV(4).这些适当的斯托克斯频移和它们基态到激发态结构的微小的改变是一致的,由于受溶剂极性的影响斯托克斯频移的大小略有不同,但差别不大.
3 结 论
从理论上研究了配合物[Os(DMSO)2(CN)2(N^N)]在四种不同溶剂中的几何结构和光谱特征.计算结果表明由于CN基的存在,配合物分子受溶剂极性影响较大.溶剂的极性影响配合物的几何结构,在极性溶剂作用下使DMSO和CN基配体的电子云密度下降,N^N配体的电子云密度上升,从而改变了金属和各个配体的作用强弱,气态时OS-N3键长为2.128Å,而在甲醇中OS-N3键长变为2.089Å.溶剂的极性影响配合物的前线分子轨道能量,从而改变了配合物最大吸收和发射波长数值.极性溶剂使分子的HOMO轨道能显著升高,但对LUMO轨道能影响较小,从而增加了HOMO-LUMO能级间隙,改变配合物的发光颜色,气态时配合物发红光,而在水中发黄绿色光,最大发射波长蓝移44 nm.这与我们之前研究的[Os(PH3)2(CN)2(N^N)]配合物相比,配合物含有PH3配体比DMSO配体对溶剂极性更加敏感,具体的原因还有待于进一步研究.
[1] Laine P P,Bedioui F,Loiseau,et al.Conformationally gated photoinduced processes with-in photosensitizer acceptor dyads based on osmium(II)complexes with triarylpyridinio-functionalized terpyridylligands:insights from experimental study[J].J.Am.Chem.Soc.,2006,128:7510-7521.
[2] Jeffrey H P.Theoretical studies of the ground and excited electronic states in cyclometalated Phenylpyridine Ir(III)complexes using density functional theory[J].J.Phys.Chem.A,2002,106:1634-1641.
[3] Lai SW,Chan Q K W,Zhu N Y,et al.cis-Dicyanoosmium(II)diimine complexes bearing phosphine or sulfoxide ligands:spectroscopic and luminescent studies[J].Inorg.Chem.,2007,46:11003-11016.
[4] Timpson C J,Bignozzi C A,Sullivan B P,et al.Influence of solvent on the spectroscopic properties of Cyano complexes of Ruthenium(II)[J].J.Phys.Chem.,1996,100:2915-2925.
[5] Burgess J.Solvent effects on visible absorption spectra of Bis-(2,2'-bipyridyl)biscyano-iron(II),Bis-(1,10-phenanthroline)biscyano-iron(II),and related compounds[J].Spectrochim.Acta,1970,26:1369-1374.
[6] 张建坡,金丽.取代基效应对锇羟基喹啉配合物结构和光谱性质的影响[J].吉林化工学院学报,2011,28(5):26-30.
[7] Adamo C,Barone C V.Toward reliable density functional methods without adjustable parameters:The PBE0 model[J].J.Chem.Phys.,1999,110:6158-6170.
[8] Hay P J,Hay W R.Ab initio effective core potentials for molecular calculations.Potentials for K to Au including the outermost core orbitals[J].J.Chem.Phys.,1985,82:299-310.
[9] Frisch M J,Trucks G W,Schlegel H B,et al.Gaussian 03[CP].Revision C.02,Gaussian,Inc.:Wallingford,CT,2004.
