亚位点+1处突变提高软化类芽胞杆菌环糊精糖基转移酶底物麦芽糊精特异性
2014-10-31许乔艳韩瑞枝李江华堵国成刘龙陈坚
许乔艳,韩瑞枝,李江华,堵国成,刘龙,陈坚
1 江南大学 糖化学与生物技术教育部重点实验室,江苏 无锡 214122
2 江南大学 工业生物技术教育部重点实验室,江苏 无锡 214122
3 江南大学 粮食发酵工艺及技术国家工程实验室,江苏 无锡 214122
维生素C (Vitamin C, VC),又称L-抗坏血酸 (L-Ascorbic Acid, L-AA),是一种水溶性维生素。VC参与很多体内生理活动,在保持和促进人体健康以及动物生长方面具有重要的作用[1-3]。VC用途很广泛,可以作为酸味剂、还原剂、抗氧化剂、漂白剂和稳定剂,广泛应用于化妆品、食品和医药等行业中[4]。但VC本身极不稳定,位于C2位置上的羟基很容易受pH、热及Cu2+、Fe2+影响而发生氧化还原反应,导致其生理活性迅速减弱甚至消失,使其在应用上受到了很大的限制[5-7]。因此,为了提高VC的稳定性,许多VC衍生物相继出现,主要包括:VC金属盐,VC酯类以及糖基化VC[8-9]。由于VC糖基衍生物具有安全、稳定性强、在体内易降解产生L-AA等优点,能更好地为人体和动物吸收和利用,因而更具优势。在所有的VC糖基衍生物中,以 2-O-D-吡喃葡糖基-L-抗坏血酸(AA-2G)研究最多,应用最为广泛,成为最理想的VC衍生物[7]。
AA-2G是利用糖基转移酶特异性的转糖基作用将糖基供体上的葡萄糖苷转移到VC的C2上合成的。糖基转移酶的选择与使用是整个AA-2G合成中的重要环节,直接影响到AA-2G的合成效率[10]。目前报道过的糖基转移酶有:α-葡萄糖苷酶[11]、环糊精葡萄糖基转移酶 (CGT酶)[12]、淀粉酶[13]、蔗糖磷酸化酶[14]和 α-异麦芽糖基-吡喃葡糖形成酶[15]。其中来源于软化类芽胞杆菌Paenibacillus macerans的环糊精葡萄糖基转移酶 (CGT酶(EC2.4.1.19))因其催化生产 AA-2G的高产物特异性而被认为是最优选择[16]。CGT酶是一种多功能型酶,能催化3种转糖基反应 (歧化、环化和耦合反应)和水解反应[9,17]。它可以将一个或多个糖基供体环上的糖残基α-1, 2糖苷键接于L-AA的C2原子上进行修饰得到 AA-2G(n)(“n”代表连接到L-AA上的糖基数量),再由糖化酶的作用水解成AA-2G[18]。
研究发现以环糊精为糖基供体时,产物专一性比较高,但是以α-环糊精为糖基供体,成本太高;以β-环糊精为糖基供体,由于β-环糊精的溶解度较低,酶促反应效率受到较大限制,均不适用于 AA-2G大规模工业化生产[16,19]。因此,以价格低廉且易溶的麦芽糊精为糖基供体高度专一性地合成AA-2G具有重要意义。X-射线衍射研究表明 CGT酶表面存在一个明显的底物结合凹槽,在底物结合凹槽中存在9个亚位点,标记为+2~–7,每个亚位点能结合一个葡萄糖残基[20]。在之前的研究中,我们已经对–3亚位点附近的氨基酸残基进行定点突变并获得较理想的结果,说明–3亚位点是改善CGT酶对麦芽糊精的底物特异性的有效位点[21]。前期研究发现+1亚位点亦是CGT酶催化转糖基的关键位点[22]。本研究中,我们对+1亚位点附近涉及的氨基酸残基:Leu194、Ala230和His233进行定点饱和突变,旨在考察亚位点+1处突变对CGT酶以麦芽糊精为糖基供体特异性合成AA-2G的影响。
1 材料与方法
1.1 材料
1.1.1 菌株与质粒
P. macerans JFB05-01系本研究室从淀粉生产企业附近的土壤中筛选获得,菌种保藏在国家典型微生物保藏中心 (保藏号:CCTCC M203062);克隆宿主菌E. coli JM109和表达宿主菌E. coli BL21(DE3)由本实验室保藏。克隆质粒 pMD19T-simple和表达质粒 pET-20b(+)购自大连宝生物工程有限公司;重组质粒pET-20b(+)/cgt由本实验室构建并保藏(GenBank Accession No. AF047363)。
1.1.2 酶和试剂
质粒小量提取试剂盒、氨苄青霉素及麦芽糊精 (DE15-20)购自上海生工生物工程公司,琼脂糖凝胶DNA回收试剂盒、定点突变试剂盒、PCR 产物纯化试剂盒、限制性内切酶、T4 DNA连接酶、Taq酶、PrimerStarDNA聚合酶购自大连宝生物工程有限公司。AA-2G购自日本林原生化研究所,L-AA购自江苏江山制药有限公司,其他试剂均为国产分析纯。
1.1.3 培养基
培养基LB、TB均按Invitrogen公司操作手册方法配制。
1.2 方法
1.2.1 突变质粒的构建与转化
以质粒pET-20b(+)/cgt为模板,设计引物,采用突变试剂盒一步PCR法对CGT酶 +1亚位点附近的氨基酸残基 (Leu194、Ala230、His233)进行定点饱和突变。以第一轮突变的产物为模板进行两点和三点复合突变。Leu194、Ala230、His233定点饱和突变及复合突变的引物见表1。

表1 定点突变引物Table 1 Primers used for site-directed mutagenesis
PCR产物纯化后,经定点突变试剂盒处理转化E. coli JM109,挑取阳性克隆,送上海生物工程有限公司测序。将鉴定正确的突变质粒分别转化表达宿主E. coli BL21 (DE3),得到含有突变质粒的基因工程菌。
1.2.2 突变体的生产与纯化
种子培养:将含突变质粒的 E. coli BL21(DE3)接入装有20 mL LB培养基的250 mL三角瓶中,回旋式摇床转速200 r/min,培养温度为37 ℃,培养8 h。
发酵培养:将培养好的种子培养液按4% (V/V)的接种量,接种至装有100 mL TB培养基的500 mL三角瓶中进行培养,开始培养温度为 37 ℃,摇床转速 200 r/min,当菌体培养至OD600为0.6时,添加IPTG至0.01 mmol/L,迅速转至25 ℃摇床,继续诱导90 h。
各培养基使用前添加100 μg/mL氨苄青霉素。
含有突变质粒的基因工程菌培养90 h后的发酵液在8 000 r/min、4 ℃下离心15 min,除去菌体,得上清即粗酶液。所有饱和突变体粗酶液的纯化均采用Ni柱一步亲和层析法[5]。
1.2.3 AA-2G的合成与检测
将纯化后的酶液用醋酸-醋酸钠缓冲液(pH 5.5)稀释至蛋白浓度为1 mg/mL,取适量与底物麦芽糊精和L-AA (5%, W/V)混合,以2 mL甘油管作为反应容器,基本装满,黑色纸包裹避光避氧下37 ℃反应24 h,然后加入10 U/mL糖化酶在65 ℃、pH 5.5条件下反应24 h,通过HPLC方法检测AA-2G产量。
HPLC检测AA-2G产量方法:酶反应样品通过0.22 μm滤膜过滤,使用Amethyst C18-H柱 (4.6 mm×250 mm, Sepax, America)检测。检测波长:238 nm;流动相:0.05 mol/L KH2PO4/H3PO4(pH 2.0);流速:0.6 mL/min。在此条件下,在大约10 min时会出现AA-2G的流出峰。AA-2G浓度通过峰面积计算而得。
在初始转化条件 (37 ℃,pH 5.5)的基础上,考虑不同反应温度(20 ℃、28 ℃、36 ℃、44 ℃和52 ℃)和 pH (醋酸-醋酸钠缓冲液:pH 4.0、4.5、5.0、5.5和6.0;磷酸缓冲液:pH 6.0、6.5、7.0和8.0)对野生型/突变型CGT酶合成AA-2G的影响。
将野生型(突变型)CGT酶与不同浓度的麦芽糊精(0.23、0.46、1.16、2.3、11.6和23.2 mmol/L)和 L-AA (2.83、5.67、28.3、56.7 和 141.5 mmol/L)混合反应测定AA-2G合成的动力学参数。采用SigmaPlot (Jandel Scientific)拟合数据,得到下面的公式:
乒乓(Ping-Pong)反应机制:
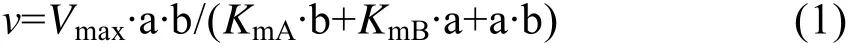
底物抑制反应机制:
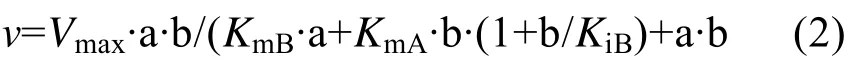
其中:v为不同底物浓度下的反应速率 (每毫克酶每小时催化生成AA-2G的量,mmol/(L·mg·h));Vmax为反应的最大速率(mmol/L·mg·h);a 和 b 分别为糖基供体 (麦芽糊精)和受体 (L-AA)的浓度 (mmol/L);KmA和KmB分别为底物麦芽糊精和L-AA的亲和常数;KiB为底物L-AA的抑制常数。
1.2.4 酶活测定
甲基橙法测定 α-环化活力的方法[23]:取适当稀释的酶液0.1 mL,加入装有0.9 mL预先用50 mmol/L磷酸缓冲液 (pH 6.5)配制的3%可溶性淀粉溶液中,在40 ℃下反应10 min后,加入1.0 mL 1.0 mol/L的盐酸停止反应,再加入1.0 mL用 50 mmol/L磷酸缓冲液配制的0.1 mmol/L甲基橙,在16 ℃下保温20 min,在505 nm下测定吸光度。一个酶活单位定义在该条件下每分钟生成1 μmol α-环糊精所需酶量。
淀粉水解活力测定方法[9]:将适量的酶液加入到含2%可溶性淀粉的50 mmol/L磷酸缓冲液中 (pH 6.5),50 ℃反应10 min,然后用DNS法测定还原糖浓度。一个酶活单位定义在该条件下每分钟生成1 μmol还原糖所需酶量。
歧化反应活力测定方法[9]:将含有6 mmol/L供体底物 4-硝基苯基-α-D-麦芽庚糖-4-6-O-亚乙基 (EPS)和 10 mmol/L受体底物麦芽糖的10 mmol/L柠檬酸缓冲液 (pH 6.0)在50 ℃保温10 min中,然后加入适当稀释的酶液0.1 mL反应,每0.5 min 取100 μL 反应样品加入20 μL 1.2 mol/L HCl (4 ℃),然后在60 ℃保温10 min使CGTase失活。随后,加入 20 μL 1.2 mol/L NaOH中和,将样品加到磷酸缓冲液 (pH 7.0),并加入 60 μL (1 U)α-糖苷酶于 37 ℃反应60 min。加入1 mL 1 mol/L碳酸钠使样品pH升至 8以上,在 401 nm波长下侧吸光值(ε401=18.4 mmol/L)。1单位酶活定义为每分钟转化1 μmol的酶的量。
1.2.5 突变体的晶体结构模拟
野生型 (突变型)CGT酶的理论晶体结构通过SWISS-MODEL蛋白模拟在线服务器 (http://www.expasy.ch/swissmod/SWISS-MODEL.html[24])进行同源模拟获得,模板为来源于B. circulans 251的CGT酶 (PDB编码:1CXK)[25]。理论结构与模板具有68.4%的同源性,序列对比显示仅在249位的氨基酸存在差异。所有分子结构图形均由Accelrys Discovery Studio Client 2.5软件制作。通过组合扩展方法 (http://cl.sdsc.edu/[26])进行结构比对,对模型进行PROCHECK[27]、Verify3D[28]和ProQ[29]分析,发现有 95%的残基位于理想区域,仅 0.2%位于未知区域。模型与模板之间的根均方偏差 (RMSD)也由组合扩展方法[26]计算而得,发现模型与模板的外观结构十分相似 (通过Cα原子最小二乘法原则进行叠加,RMSD为0.5 Å)。麦芽九糖抑制剂由模板PDB 1CXK的活性位点转移至模型的活性位点。最后,酶-底物反应的能量由Accelrys Discovery Studio Client 2.5提供的Amber-based能量最小化方法计算获得。
2 结果与分析
2.1 突变体的AA-2G合成
所有的突变体均由定点突变技术成功构建并经DNA测序鉴定正确。将野生型和饱和突变获得的所有突变型CGT酶经E. coli BL21 (DE3)表达,SDS-PAGE鉴定发现表达水平及分子量均无明显变化。粗酶液经Ni-柱纯化,得到电泳纯蛋白。
在构建的所有单点突变体中,突变体L194N、A230D、H233E相较于野生型CGT酶产AA-2G的量最高 (其他单点突变体产 AA-2G的量均低于野生型CGT酶或提高率在10%以下)。在这3株优势突变体的基础上,我们再进行两点和三点复合突变,得到L194N/A230D、L194N/H233E、A230D/H233E和L194N/A230D/H233E四株复合突变体。其中,三点复合突变体L194N/A230D/H233E以麦芽糊精为糖基供体产AA-2G的量最高为1.95 g/L,较野生型提高了62.5% (表2)。
2.2 突变体酶法合成 AA-2G的最适温度和最适pH
野生型 (突变型)CGT酶以麦芽糊精为糖基供体合成AA-2G的最适反应温度为36 ℃,与α-CGT酶催化以β-环糊精为底物合成AA-2G的最适反应温度一致[18],而重组α-CGT酶催化环化反应的最适温度为45 ℃[23]。在20–52 ℃的范围内,野生型CGT酶催化合成AA-2G的能力变化不大。当温度升高至44 ℃时,突变体产AA-2G的量下降较快,52 ℃时突变体催化合成AA-2G的产量降至36 ℃时的一半左右 (图1)。
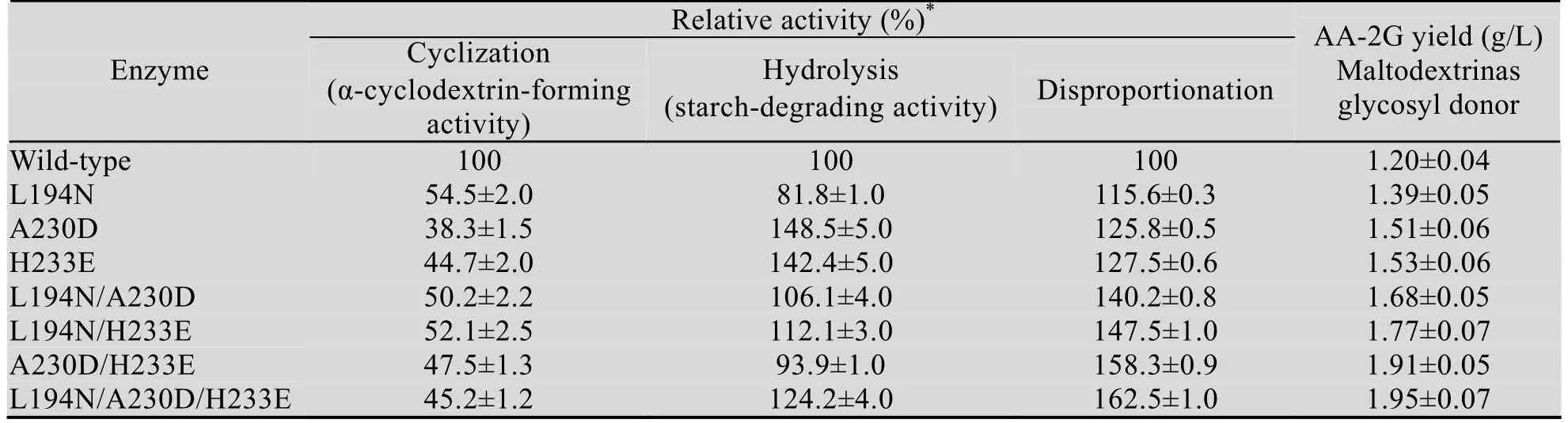
表2 野生型 (突变型)CGT酶的相对酶活和AA-2G产量Table 2 Comparison of the reaction activities and AA-2G yields of the wild-type and mutant CGTases

图1 反应温度对野生型(突变型)CGT酶以麦芽糊精为糖基供体合成AA-2G的影响Fig. 1 Effect of reaction temperature on AA-2G synthesis by the wild-type and mutant CGTases with maltodextrin as the glycosyl donor.
图 2反映了不同 pH对野生型 (突变型)CGT酶催化合成 AA-2G的影响。与野生型相比,突变体的最适反应 pH有一定程度上的变化。其中,突变体L194N、L194N/A230D与野生型一致,催化合成AA-2G的最适pH为6.5;而突变体 A230D、H233E、L194N/H233E、A230D/H233E和 L194N/A230D/H233E则在pH 7.0时获得最高的AA-2G产量。初步推测,突变体引入的不同极性的氨基酸可能改变了底物结合活性位点可解离基团的 pKa,进而造成最适反应pH的改变。
图3为在最适温度和最适pH条件下,野生型和突变型CGT酶催化合成AA-2G产量随时间的变化曲线。在反应的初始阶段,AA-2G的产量有明显的增长,野生型和突变体均在 24 h时获得AA-2G 的最高产量 (表 2)。在所有突变体中,L194N/A230D/H233E的产量最高,约为野生型的1.6倍。
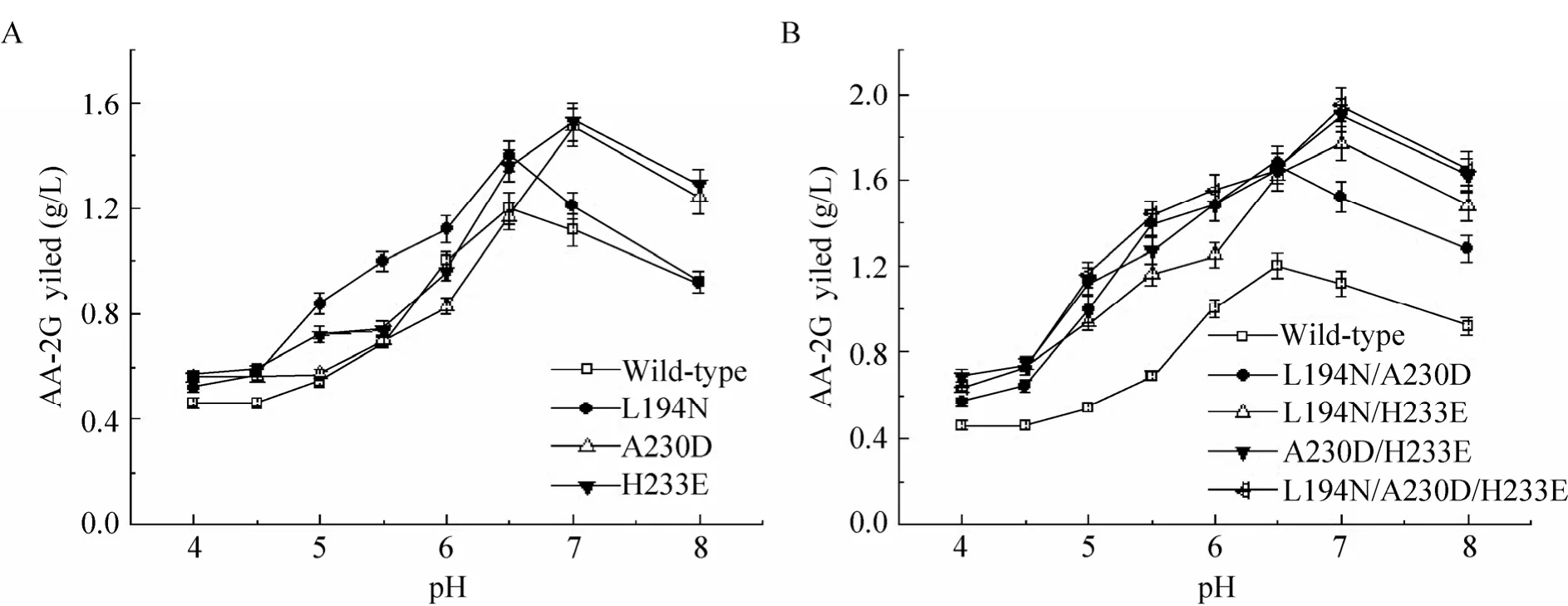
图2 反应pH对野生型 (突变型)CGT酶以麦芽糊精为糖基供体合成AA-2G的影响Fig. 2 Effect of reaction pH on AA-2G synthesis by the wild-type and mutant CGTases with maltodextrin as the glycosyl donor.

图3 野生型 (突变型)CGT酶以L-AA和麦芽糊精为底物合成AA-2G的时间产量曲线Fig. 3 Time profiles of AA-2G synthesis by the wild-type and mutant CGTases with L-AA and maltodextrin as the substrates.

图4 野生型CGT酶(a)和突变体L194N/A230D/H233E (b)合成AA-2G反应的Lineweaver-Burk曲线Fig. 4 Lineweaver-Burk plots of the AA-2G synthesis by the wild-type CGTase (A)and the mutant L194N/A230D/H233E (B). L-AA concentrations (mmol/L): ■: 2.83; ●: 5.67; ▲: 28.3; ◆: 56.7; ★: 141.5. Linear regression of the experimental data is represented by black dotted lines, and the calculated values from Equation (1)and(2)are represented by red solid lines.
2.3 突变体的反应动力学及作用机理分析
为了研究野生型 (突变型)CGT酶的动力学性质,分别测定了以不同浓度的麦芽糊精和L-AA为底物时 AA-2G的产量。拟合数据并对野生型CGT酶及突变体 L194N/A230D/H233E进行Lineweaver-Burk作图,得到图4。如图4A所示,对野生型CGT酶进行动力学分析,经双倒数线性拟合得到的直线符合多底物酶促反应的乒乓机制,与公式(1)计算的值相符合;而突变体L194N/A230D/H233E线性拟合的结果与公式(2)计算的值较符合,说明其酶促反应符合底物抑制动力学模型,高浓度的底物L-AA对AA-2G的合成存在抑制作用 (图4B)。另外几个突变体的分析结果也符合公式(2),详细的动力学参数见表3。
突变体的最大反应速率 (Vmax)要高于野生型,Km(麦芽糊精)和Kcat/Km(麦芽糊精)值的变化显示出突变体对底物麦芽糊精的亲和性和酶促反应的催化效率与野生型相比都有一定程度的提高。突变体相对于野生型 Km(L-AA)和 Kcat/Km(L-AA)值的变化则说明突变体对底物 L-AA的亲和性有所降低。抑制常数Ki(L-AA)表明高浓度的L-AA对突变体K47L/Y89F/N94P/D196Y催化的的酶促反应抑制作用最显著。
我们还考察了突变对 CGT酶 α-环化活力、淀粉水解活力及歧化活力的影响。如表2所示,突变型 CGT酶的 α-环化活力降至野生型的一半甚至更低,而除突变体L194N和A23D/H233E的淀粉水解活力较野生型分别降低了18.2%和6.1%外,大部分突变型CGT酶的淀粉水解活力均有一定程度的提高。突变体L194N、A230D、H233E、L194N/A230D、L194N/H233E、A230D/H233E和L194N/A230D/H233E的歧化反应活力较野生型CGT酶分别提高了15.6%,25.8%,27.5%,40.2%,47.5%,58.3%和62.5%。这一增长趋势与AA-2G产量的趋势一致,说明歧化反应是以麦芽糊精做为糖基供体生产AA-2G的关键反应。
图5A–D依次为野生型CGT酶和第94位亮氨酸,第230位丙氨酸和第 233位组氨酸分别被天冬酰胺、天冬氨酸和谷氨酸取代的突变体,可以看出突变体与野生型CGT酶的氨基酸侧链有很大的差别。
如图5B,CGT酶第194位的氨基酸由亮氨酸突变为天冬酰胺,使侧链结构发生了改变,进而引发了附近的氨基酸残基及整个空间结构的变化,可能在一定程度上改善了CGT酶与线性底物麦芽糊精间的结合,提高了AA-2G的合成效率。
如图5C所示,CGT酶第230位的丙氨酸突变为天冬氨酸后,与底物分子间的氢键作用力较突变前有所增强。增加的氢键使底物分子更趋向于第 230位的氨基酸残基,可能在一定程度上阻滞了线性底物的环化,造成突变体A230D环化活力的大幅度降低。然而增强的氢键作用力也可能有效改善了突变体A230D对线性底物(如麦芽糊精)的亲和性,更利于其以麦芽糊精为糖基供体合成更高产量的 AA-2G。突变体H233E底物特异性改善的机理与突变体A230D类似 (图 5D)。相关动力学参数 Km(麦芽糊精)和Kcat/Km(麦芽糊精)的变化也证实了这一点。
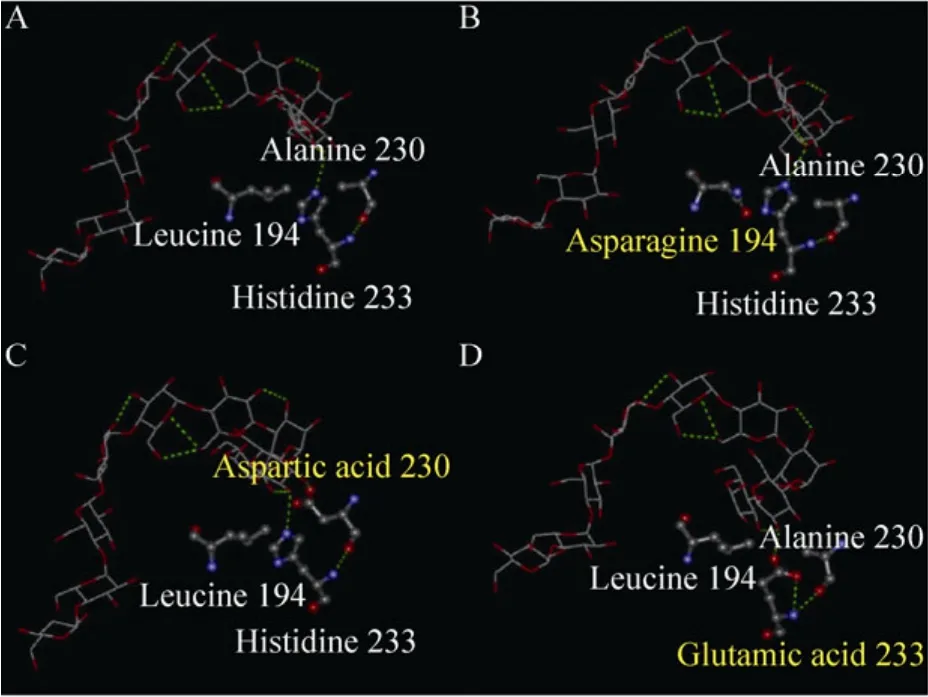
图5 野生型 (突变型)CGT酶与麦芽九糖抑制剂在+1亚位点上作用的模拟结构图Fig. 5 Close-up the wild-type and mutant CGTases theoretical structure with a maltononaose substrate at subsite +1 (PDB accession code 1CXK). (A)Wild-type CGTase. (B)L194N. (C)A230D. (D)H233E.

表3 野生型 (突变型)CGT酶的动力学参数Table 3 Kinetic parameters of the wild-type and mutant CGTases
3 结论
本实验通过定点突变技术构建了 7株底物麦芽糊精特异性明显改善的CGT酶突变体,其中三点突变体L194N/A230D/H233E以麦芽糊精为底物合成AA-2G的产量最高为1.95 g/L,较野生型CGT酶提高62.5%。通过动力学分析和晶体结构模拟我们初步阐述了突变体底物麦芽糊精特异性改善的机理。研究结果使我们进一步了解了+1亚位点附近氨基酸残基的突变能有效提高CGT酶与线性底物分子的亲和性。为了进一步提高AA-2G的产量,还需继续开展CGT酶的固定化和转化条件优化等工作。
[1]Englard S, Seifter S. The biochemical functions of ascorbic acid. Annu Rev Nutr, 1986, 6(1):365–406.
[2]Jacob RA, Sotoudeh G. Vitamin C function and status in chronic disease. Nutri Clin Care, 2002,5(2): 66–74.
[3]Naidu KA. Vitamin C in human health and disease is still a mystery? An overview. Nutr J, 2003, 2(1): 7.
[4]Fujiwara S, Kakihara H, Sakaguchi K, et al.Analysis of mutations in cyclodextrin glucanotransferase from Bacillus stearothermophilus which affect cyclization characteristics and thermostability. J Bacteriol,1992, 174(22): 7478–7481.
[5]Li ZF, Zhang JY, Sun Q, et al. Mutations of lysine 47 in cyclodextrin glycosyltransferase from Paenibacillus macerans enhance β-cyclodextrin specificity. J Agric Food Chem, 2009, 57(18):8386–8391.
[6]Penninga D, Strokopytov B, Rozeboom HJ, et al.Site-directed mutations in tyrosine 195 of cyclodextrin glycosyltransferase from Bacillus circulans strain 251 affect activity and product specificity. Biochem, 1995, 34(10): 3368–3376.
[7]Yamamoto I, Muto N, Murakami K, et al.L-ascorbic acid alpha-glucoside formed by regioselective transglucosylation with rat intestinal and rice seed alpha-glucosidases: its improved stability and structure determination. Chem Pharm Bull (Tokoyo), 1990, 38(11): 3020.
[8]Sin KA, Nakamura A, Masaki H, et al.Replacement of an amino acid residue of cyclodextrin glucanotransferase of Bacillus ohbensis doubles the production of γ-cyclodextrin.J Biotechnol, 1994, 32(3): 283–288.
[9]van der Veen BA, Uitdehaag J, Penninga D, et al.Rational design of cyclodextrin glycosyltransferase from Bacillus circulans strain 251 to increase α-cyclodextrin production. J Mol Biol, 2000,296(4): 1027–1038.
[10]Han R, Liu L, Li J, et al. Functions, applications and production of 2-O-d-glucopyranosyl-l-ascorbic acid.Appl Microbiol Biotechnol, 2012, 95: 313–320.
[11]Muto N, Suga S, Fujii K, et al. Formation of a stable ascorbic acid 2-glucoside by specific transglucosylation with rice seed α-glucosidase.Agricul Biol Chem, 1990, 54(7): 1697–1703.
[12]Aga H, Yoneyama M, Sakai S, et al. Synthesis of 2-O-α-D-glucopyranosyl L-ascorbic acid by cyclomaltodextrin glucanotransferase from Bacillus stearothermophilus (Biological Chemistry). Agric Biol Chem, 1991, 55(7): 1751–1756.
[13]Lee SB, Nam K, Lee SJ, et al. Antioxidative effects of glycosyl-ascorbic acids synthesized by maltogenic amylase to reduce lipid oxidation and volatiles production in cooked chicken meat. Biosci Biotechnol Biochem, 2004, 68(1): 36–43.
[14]Kwon T, Kim CT, Lee JH. Transglucosylation of ascorbic acid to ascorbic acid 2-glucoside by a recombinant sucrose phosphorylase from Bifidobacterium longum. Biotechnol Lett, 2007, 29(4):611–615.
[15]Mukai K, Tsusaki K, Kubota M, et al. Process for producing 2-O-alpha-D-glucopyranosyl-L-ascorbic acid: EP patent, 1553186. 2005-07-13.
[16]Jun HK, Bae KM, Kim SK. Production of 2-O-α-D-glucopyranosyl L-ascorbic acid using cyclodextrin glucanotransferase from Paenibacillus sp.. Biotechnol Lett, 2001, 23(21): 1793–1797.
[17]Henrissat B. A classification of glycosyl hydrolases based on amino acid sequence similarities.Biochem J, 1991, 280(2): 309.
[18]Zhang Z, Li J, Liu L, et al. Enzymatic transformation of 2-O-α-D-glucopyranosyl-L-ascorbic acid by α-cyclodextrin glucanotransferase from recombinant Escherichia coli. Biotechnol Bioproc E, 2011, 16(1):107–113.
[19]Tanaka M, Muto N, Yamamoto I. Characterization of Bacillus stearothermophilus cyclodextrin glucanotransferase in ascorbic acid 2-O-α-glucoside formation. Biochim Biophys Acta, 1991, 1078(2):127–132.
[20]Strokopytov B, Knegtel RMA, Penninga D, et al.Structure of cyclodextrin glycosyltransferase complexed with a maltononaose inhibitor at 2.6 Å resolution. Implications for product specificity.Biochemistry, 1996, 35(13): 4241–4249.
[21]Liu L, Xu Q, Han R, et al. Improving maltodextrin specif i city for enzymatic synthesis of 2-O-d-glucopyranosyl-l-ascorbic acid by site-saturation engineering of subsite-3 in cyclodextrin glycosyltransferase from Paenibacillus macerans. J Biotechnol, 2013, 166: 198–205.
[22]Leemhuis H, Rozeboom HJ, Wilbrink M, et al.Conversion of cyclodextrin glycosyltransferase into a starch hydrolase by directed evolution: the role of alanine 230 in acceptor subsite +1. Biochem, 2003,42(24): 7518–7526.
[23]Li Z, Li B, Gu Z, et al. Extracellular expression and biochemical characterization of α-cyclodextrin glycosyltransferase from Paenibacillus macerans.Carbohyd Res, 2010, 345(7): 886–892.
[24]Arnold K, Bordoli L, Kopp J, et al. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics, 2006, 22(2): 195–201.
[25]Uitdehaag JCM, Mosi R, Kalk KH, et al. X-ray structures along the reaction pathway of cyclodextrin glycosyltransferase elucidate catalysis in the α-amylase family. Nat Struct Biol, 1999, 6(5):432–436.
[26]Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension(CE)of the optimal path. Protein Eng, 1998, 11(9):739–747.
[27]Laskowski RA, MacArthur MW, Moss DS, et al.PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst, 1993, 26(2): 283–291.
[28]Bowie JU, Luthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science, 1991,253(5016): 164–170.
[29]Wallner B, Elofsson A. Can correct protein models be identified? Protein Sci, 2009, 12(5): 1073–1086.
