亚热带土壤氮素反硝化气态产物研究
2014-10-22续勇波蔡祖聪
续勇波,蔡祖聪
1.云南农业大学烟草学院,云南 昆明 650201;2.南京师范大学地理科学学院,江苏 南京 210097
反硝化是指将硝态氮或亚硝态氮还原成一氧化氮(NO)、一氧化二氮(N2O)和氮气(N2)的过程,是氮循环中重要的转化过程和环节。反硝化作用的环境效应取决于其所产生的终产物,以及不同产物间的比例。通常认为反硝化作用的主要终产物是N2O和N2,NO并不是土壤或水体中反硝化作用的主要终产物。但这是基于温带恒电荷土壤的研究结果(Hofstra和Bouwman, 2005)。关于热带亚热带土壤反硝化过程中NO排放的研究甚少(Maljanen等, 2007),因此,影响热带亚热带地区土壤反硝化产物的主要因素和机理尚不十分清楚。
热带亚热带土壤为可变电荷土壤,土壤的化学性质与温带地区恒电荷土壤有诸多不同特点(徐仁扣等, 2014),如土壤高度风化、铁铝氧化物含量显著高于温带地区土壤(Qafoku等,2004;Zhang等,2009),土壤氧化还原势较高(丁昌璞,2008),酸性较强(Xu和Cai,2007),为继承性肥力较低的氧化土(Oxisols)和老成土(Ultisols)。越来越多的证据表明热带亚热带土壤反硝化具有一些与温带土壤不同的特征(Zhang等,2009 ;Xu和 Cai,2007; Zhang等,2010),然而还需要进一步深入研究造成热带亚热带土壤和温带土壤反硝化特性不同的原因和机理,这将有助于加深对热带亚热带环境条件下土壤 N循环的理解和认识。
在前期的研究工作中,我们对采集自亚热带不同成土母质和利用方式的45个土壤样本,在密闭淹水厌氧培养试验条件下,研究了亚热带土壤反硝化特性、影响因素和机理等(Xu和Cai,2007;Xu等,2012),发现随着培养时间的延长,排放到培养瓶上部空间的 N2O浓度逐渐上升,在第3~8天达到高峰,之后由于N2O被土壤进一步还原成为N2而逐渐降低(Xu等,2012)。那么在达到N2O浓度高峰之前是否存在N2O产生的前提物质——NO的大量产生?为此,我们进行了稳定性同位素15N示踪试验,用K15NO3替代前述试验中的普通 KNO3,研究厌氧培养 1周内反硝化过程中的产物问题。

表1 供试土样理化性质Table 1 Selected physico-chemical properties of the tested soils
1 材料和方法
1.1 供试土样和试验方法
供试土样采自具有典型亚热带季风湿润性气候 特 点 的 江 西 省 ( 24°29'~30°04'N,113°34'~118°28'E)。由于受东南季风气候的影响,本区气候高温多雨,干湿季季节明显。根据近 30年气象统计资料,其年平均降雨量为1785 mm,其中50%集中于4—6月,年平均温度为18.4 ℃,最高月平均温29.9 ℃出现于7月,无霜期为261 d,≥10 ℃的积温为 5000~9500 ℃。土壤类型为低活性强酸土(Acrisols)和铁铝土(Ferralsols)(FAO,2001)。供试土样选择包括由3种成土母质(即第四纪红土(Q)、第三纪红砂岩(S)和花岗岩(G))和5种利用方式(即林地(F)、灌丛(B)、茶园(T)、旱地(U)和稻田(R))组成的6个土壤类型组合,即SR、QR、QU、QB、GT、GF,采集0~20 cm新鲜土样,风干1~3 d,以能通过2 mm筛为宜,置于4 ℃保存备用并立即开始试验。供试土壤理化性质见表1。
试验方法参照文献(Xu和Cai,2007;Xu等,2012),为了测定同位素15N丰度,采用K15NO3替代普通 KNO3,试验步骤和测试方法稍作改动,具体为称取相当于 30 g烘干土质量的上述新鲜土装入250 mL三角瓶中,加蒸馏水30mL,盖上橡胶塞,于30 ℃下淹水预培养7 d,以便激活土壤微生物。同时测定培养瓶有效空间体积和土壤全氮、NH4+-N,NO3--N的质量分数,作为土壤背景值。预培养结束时,随机选取3瓶测定土壤全氮、NH4+-N、NO3--N的质量分数,记做起始质量分数。
将三角瓶用带采气孔的特殊硅橡胶塞(硅橡胶塞中间打一小孔,内插玻璃管,管外套一段细硅橡胶管,其口用硅橡胶塞封口)盖紧,并在瓶塞周围涂抹704胶密封以防漏气。从取样口将三角瓶内抽成真空,通人氮气,再抽成真空,如此反复3次,最后通入1个大气压的高纯氮。用注射器加入5 mL(含6 mg NO3--N)硝酸钾溶液(K15NO3, 10 atom%15N)(相当于200 mg·kg-1),并用5 mL蒸馏水清洗1次,充分混合。最后将取样口用704胶密封以保证厌氧培养环境,所有三角瓶置于 30 ℃下恒温淹水厌氧培养。
分别在厌氧培养开始后的1、3、7 d采集20 mL气样3份,分别注入18 mL已抽成真空的贮气瓶中,第1瓶供测定N2O体积分数,第2瓶供测定15N2丰度,第3瓶供测定15N2O丰度用,抽取气样前用注射器反复抽提瓶内气体3次以混匀气体。取完气样后按V(液)∶m(土)=4∶1 加入 3 mol·L-1KCl 80 mL(已加水量考虑在内,使最终浓度为2 mol·L-1,总体积为 120 mL),于 25 ℃、250 r·min-1下震荡 1 h,离心,定量滤纸过滤,收集滤液于塑料瓶中。
吸取一定体积的滤液于蒸馏装置中,加入0.2 g氧化镁蒸馏,以硫酸标准溶液滴定,测定馏出液中NH4+-N的质量分数;再加代氏合金蒸馏,转化成铵盐后, 硫酸标准溶液滴定,测定(NO3-+NO2-)-N总量。在测定完硝态氮和氨态氮质量分数的溶液中分别加入6滴硫酸溶液(3 mol·L-1)酸化,于80 ℃烘箱中浓缩至干,用同位素质谱仪分析丰度。
离心管中的土壤转移到铝盒中,放入 45 ℃烘箱中烘干,磨细过100目筛,称取适量土样于消煮管中,按土壤全氮开氏消煮法进行消煮、蒸馏、硫酸滴定,测定完全氮质量分数后,滴定过的馏出液同上述方法酸化,放入 80 ℃烘箱中浓缩至干,用同位素质谱仪分析土壤全氮中的15N丰度。同时测定土壤含水量。
1.2 计算公式
N2O气体体积分数用装有63Ni电子捕获器(ECD)的气相色谱仪 (Shimadzu GC-14B, 日本)测定。色谱柱为80/100目的Porapak Q填充柱,进样口、检测器以及填充柱的温度分别为100 ℃、300 ℃和65 ℃。载气为95%氩气+5%甲烷,流速为40 mL·min-1。标准气为日本国立农业环境研究所提供。溶解到水中的N2O体积分数经Bunsen常数校正(Tiedje, 1982)。
N2O累积排放量计算公式

式中:MN为N2O的累积排放量,以N2O-N计算,单位:mg·kg-1。ρ为标准状态下N2O-N的密度,为1.25 kg·m-3;C为N2O气体体积分数,为×10-6;Vg是培养瓶上部有效空间体积,单位是m3;Vl为液体体积,单位是m3;α为Bunsen校正系数,30 ℃时为0.472;W为培养瓶内的烘干土质量,单位是kg;t为测定气体时的温度(℃)。
N2体积分数由于试验条件所限无法直接测定,通过下列公式计算得到。溶解到水中的 N2体积分数经Bunsen系数校正(Tiedje, 1982)。
在已知有效空间体积、加入的水体积、土样质量,且充满纯氮气的培养瓶中,N2累积排放量(mg·kg-1)可按下式计算:

式中:C为N2累积排放量,单位为mg·kg-1;1.13为30 ℃时的N2密度,单位为kg·m-3;Vg为培养瓶有效空间体积,单位为 m3;Vl为加入的水体积,单位为m3;a为Bunsen校正系数,30 ℃时为0.0136;W为土样质量,单位为kg。

其中:C’为15N2累积排放量,单位为mg·kg-1,C为公式(2)计算得到的培养瓶中N2累积排放量,单位为mg·kg-1;A为测定得到的15N2丰度,单位为atom%。
本文中气态氮产物的损失率是指占施入15N总量的体积分数(%),总气态氮损失率的实测值是指测定和计算得到的N2O与N2损失率的总和;总气态氮损失率的估计值为差减法计算得到,即总加入15N量减去土壤全氮、铵态氮和硝态氮中的15N的差值。
2 结果与分析
从表2中不同取样时间观测数据可以看出,随培养时间延长,土壤15NO3-质量分数、15N2O和15N2累积排放量有明显变化,而土壤全氮(Total15N)和15NH4+质量分数变化不大,表明该体系中发生的主要氮素转化过程是反硝化。
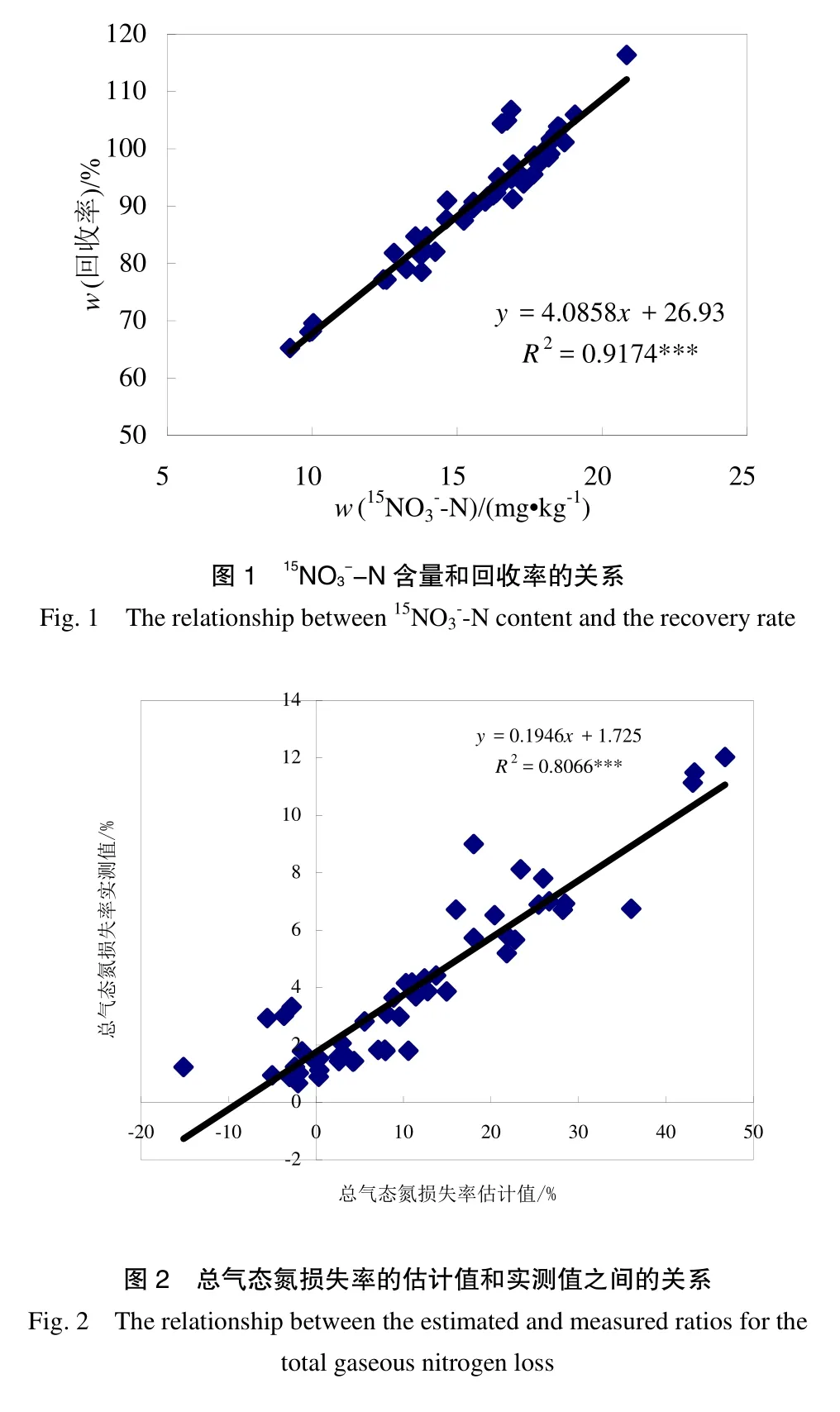
不同土样第1天的回收率都在95%以上(表2),表明测定方法和计算方法无误,误差在允许范围内。但随着培养时间的推移,回收率逐渐下降,到第 7天回收率仅为 67.2%~93.3%,平均为(80.8±9.1)%。与之相对应的是15NO3-质量分数由于反硝化作用而随培养时间逐渐减少。土壤总残留的15NO3-质量分数和回收率之间存在显著正相关关系(图1),r2=0.917(p<0.001),表明反硝化势越低的土样回收率越高。
15N2O和15N2累积排放量随培养时间推移而逐渐增加,总气态氮损失率的估计值和实测值都随培养时间延长呈上升趋势,两者之间存在显著正相关性(r2=0.81,p<0.001)(图2)。总气态氮损失率的估计值显著高于实测值(p<0.01),两者之差高达6.7%~32.8%,平均为(18.6±9.0)%,即这部分气态氮损失占施入15N总量的18.6%。而N2O、N2分别占总施入氮量的(4.4±2.3%)、(2.0±1.2)%。

表2 不同土样不同培养时间的回收率和气态氮损失Table 2 The recovery rate and the gaseous nitrogen loss for different soils at different sampling time
3 讨论
在用标准(NH4++NO3-)-N溶液对测定NO3--N和NH4+-N质量分数的氧化镁-代氏合金(MgO-Devarda)蒸馏法所做的方法检验试验结果表明,回收率均在97%以上。开氏消煮法也是成熟的土壤全氮测定方法,因此,土壤中氮的测定误差不是造成反硝化试验中总回收率低的主要原因。此外,氮化合物和N2的丰度测定方法以及 N2O体积分数的测定方法也都是成熟技术,N2累积排放量计算方法在理论上也是正确的。
尽管厌氧条件下加入的 NO3--N有可能通过非生物固定作用转化成可溶性有机氮(DON),这一N转化途径在温带森林土壤中是氮循环的重要组成环节,但在氧化还原势(Eh)较高的热带亚热带土壤中并非如此(Zhang等,2010)。因此在本试验条件下这一转化过程可忽略不计。
(N2+N2O)-15N实测值低于总气态氮损失的估计值,原因之一可能是由于对N2O和N2的不完全回收。尽管计算N2O和N2产生量已包括了溶解在水中的N2O和N2量(Tiedje, 1982),本实验按文献中的溶解系数计算得到的水中溶解的15N2O和15N2量分别占施入15N总量最高不到0.5%和0.01%,这虽然与 Buresh和 Austin(1988)提出的水中溶解的15N2O量占施入15N总量不到1%的结果相似,但有可能低估了N2O和N2产生量,因为土壤和土壤溶液均可被N2O和N2所饱和,其可溶解量可高达周围空气浓度的46倍(Kaplan等,1978)。此外,土壤也可吸附N2-15N和N2O-15N,据Lindau等(1988)报道,淹水土壤中加入15N尿素后的33 d中28%被土壤以N2-15N的形态截留,在无淋洗、径流损失的情况下直接测定得到的(N2+N2O)-15N回收率在41%~73%。而Chen等(1998)认为施入15NO3-5 d后90%的气态氮产物被土壤吸附和截留。因此,按文献中的溶解系数计算得到的水中溶解的 N2O和N2的量可能低估了溶解到水中的N2O和N2的量,也意味着忽视了被土壤吸附和截留的N2O和N2数量,从而导致计算的N2O和N2产生量有可能低估了实际N2O和N2排放量。
由于本试验条件的限制,NO累积排放量无法直接测定,但从本实验结果计算,这部分气态氮损失可能占到了总气态氮损失的67.5%~78.6%,平均为(74.1±4.2)%;占施入15N总量的(18.6±9.0)%。NO浓度有可能对反硝化气态产物的贡献达到显著水平,因此有必要对热带亚热带土壤反硝化产物 NO进行深入研究。
湿润型热带亚热带土壤自养硝化作用和反硝化作用较温带地区低(Zhang等,2010a, 2009b;Xu和Cai,2007;Zhao等,2007)。有研究表明,在气候因素、土壤特性、较快的氮循环速率等综合作用下,热带亚热带森林土壤气态氮氧化物损失较温带森林土壤高,且是全球最大的N2O和NO自然排放源(Davidson和 Kingerlee, 1997; Stehfest和Bouwman, 2006)。厌氧培养条件下,湿润热带亚热带地区森林土壤反硝化过程中产生的NO占总气态氮产物的比率和 N2O占总气态氮产物的比率通常显著高于温带森林土壤(Zhang等,2009;Parsons等,1993),Zhang等认为这和热带亚热带土壤氧化物含量显著高于温带地区土壤(丁昌璞,2008)、较高的氧化还原势抑制了土壤反硝化的进行,从而使得 NO和 N2O占总气态氮产物的比率增加有关(Zhang等,2009),进一步证实本试验供试亚热带土壤NO可能是反硝化的主要产物之一。
本试验中添加的 NO3--N质量分数为 200 mg·kg-1,NO3-浓度也是影响反硝化产物的因素之一。通常认为高浓度的 NO3-或 NO2-会抑制 NO还原为N2O,即抑制N2O的产生,使NO排放量增加。如Luo等(1996)认为NO3--N质量分数>100 mg·kg-1会抑制N2O的产生。而有的学者认为NO3--N质量分数范围在 10~30 mg·kg-1即足以抑制上述过程(Cho和Mills,1979)。通常在施肥后农田土壤NO3-浓度可达这一标准。由于电子竞争作用,高浓度的NO3-不仅抑制N2O还原酶的合成和活性,而且也能抑制NO还原酶和NO2-还原酶(Gaskell等,1981)。反硝化还原酶的合成存在依次滞后的现象,即酶合成的先后顺序是NO3-还原酶、NO2-还原酶、NO还原酶和 N2O还原酶,而且当 NO3-浓度越高,合成滞后的时间越长,因此有NO、N2O的暂时积累,且NO、N2O的排放先于N2的排放(Holtan-Hartwig等,2000)。
反硝化菌的生长和酶的合成在含有丰富氧化物且氧化还原势较高的湿润型热带亚热带土壤上也可能会受到抑制(Zhang等,2009)。我们的研究结果也证实湿润型亚热带土壤反硝化菌丰富度较低(Xu和 Cai,2007)。然而关于土壤氧化物如何直接影响反硝化微生物的生长、酶合成和活性有待于进一步研究。另外,湿润热带亚热带土壤pH值较低,远远低于大多数反硝化菌生长和活性发挥的适宜pH值6~8范围(Aulakh等,1992)。
已有大量证据表明 NO也可由化学反硝化产生,即厌氧条件下 Fe2+和 NO3-或 NO2-发生化学反硝化产生Fe3+和NO(Philips等,2003)。我国亚热带土壤中含有丰富的铁氧化物(徐仁扣等,2014;Qafoku等,2004),其活性较高的部分为Fe参与反硝化创造了物质基础,我们的试验结果也证实了酸性亚热带土壤Fe2+参与了NO3-反硝化过程(Xu和Cai,2007;Xu 等,2013)。
以上这些因素的综合作用可能导致反硝化过程中N2O和N2排放量较低,而NO是反硝化的主要产物之一。这也可能是造成反硝化回收率低的原因之一。但是造成热带亚热带地区土壤和温带地区土壤反硝化气态产物及不同产物间比例差异的因素和机理尚需进一步研究。
从我们以往的研究结果可知,亚热带土壤N2O产生速率较其被进一步还原的速率高(Xu等,2012),因此,我们有理由推测反硝化在厌氧培养开始的头几天时间里,相对于N2O还原潜势较弱、N2O产生潜势较大,N2O排放占主导地位的情况下,作为N2O生成前提物质的NO有可能大量累积。如果不能回收的那部分都以NO形式排放,本实验中NO损失平均占总气态氮损失的(74.1±4.2)%,而以N2O和 N2形态损失的氮素只分别占总气态氮损失的(17.1±2.1)%、(8.7±4.8)%。NO在大气中的活性很强,是形成酸雨和光化学烟雾的前提物质(Hou等,2000),其环境危害不容低估。
此外,反硝化气态产物中NO和N2O总量占总气态氮损失的91.3%,意味着在亚热带土壤氮素反硝化中NO可能是反硝化的主要终产物,而非对环境无害的N2,因此在亚热带土壤反硝化研究中使用乙炔抑制方法可能意义不大。这一推理可从王连峰(2003)以亚热带红壤为材料所做的反硝化试验结果中得到部分支持,即经淹水或湿润(40%WHC)处理110 d的旱地红砂土在100%WHC水分条件下培育5 d,乙炔的加入与否对土壤N2O累积排放量未造成显著影响。
4 结论
1)亚热带土壤反硝化培养过程中随培养时间推移,15N回收率逐渐下降,土样反硝化作用强弱与回收率呈显著负相关。
2)总气态氮损失率的估计值和实测值都随培养时间延长呈上升趋势,两者之间存在显著正相关性。
3)根据15N示踪试验结果估计,厌氧培养 7天内反硝化作用产生的气态产物中NO、N2O和N2分别占总气态氮损失的74.1%、17.1%和8.7%。
4)反硝化作用产生的气态产物中NO、N2O和N2分别占总施入氮量的18.6%、4.4%和2.0%。
5)亚热带土壤氮素反硝化过程中 N2O和 N2排放量较低,而NO是反硝化的主要气体产物之一,其环境危害不容低估。
AULAKH M S, DORAN J W, MOSIER A R.1992.Soil denitrification -significance, measurement, and effects of management [C] //Stewart B A.Advances in Soil Science.New York: Springer-Verlag: 1-57.
BURESH R J, AUSTIN E R.1988.Direct measurement of dinitrogen and nitrous oxide flux in flooded rice fields [J].Soil Science Society of America Journal, 52: 681-688.
CHEN D L, CHALK P M, FRENEY J R.1998.Nitrogen transformations in a flooded soil in the presence and absence of rice plants: 2.Denitrification [J].Nutrient cycling in agroecosystems, 51: 269-279.
CHO C M, MILLS J G.1979.Kinetic formulation of the denitrification process in soil [J].Canadian Journal of Soil Science, 58: 443-457.
DAVIDSON E A, KINGERLEE W.1997.A global inventory of nitric oxide emissions from soils [J].Nutrient Cycling in Agroecosystems, 48:37-50.
FAO.2001.World Soil Sources Reports 94 [C] //Driessen P, Deckers J,Nachtergaele F.Lecture Notes on the Major Soils of the World.Rome:FAO: 123-125.
GASKELL I F, BLACKMER A M, BREMNER J M.1981.Comparison of effects of nitrate, nitrite and nitric oxide on reduction of nitrous oxide to dinitrogen by soil microorganisms [J].Soil Science Society of America Journal, 45: 1124-1127.
HOFSTRA N, BOUWMAN A F.2005.Denitrification in agricultural soils:summarizing published data and estimating global annual rates [J].Nutrient Cycling in Agroecosystems, 72: 267-278.
HOLTAN-HARTWIG L, DOÈRSCH P, BAKKEN L R.2000.Comparison of denitrifying communities in organic soils: kinetics of NO3-and N2O reduction [J].Soil Biology and Biochemistry, 32: 833-843.
HOU A, AKIYAMA H, NAKAJIMA Y, et al.2000.Effects of urea form and soil moisture on N2O and NO emissions from lapanese Andosols [J].Chemosphere - Global Change Science, 2: 321-327.
KAPLAN W A, ELKINS J W, KOLB C E, et al.1978.Nitrous oxide in fresh water systems: an estimate for the yield of atmospheric N2O associated with disposal of human waste [J].Pure and Applied Geophysics, 116: 423-438.
LINDAU C W, PATRICKJR W H, DELAUNE R D, et al.1988.Entrapment of nitrogen-15 dinitrogen during soil denitrification [J].Soil Science Society of America Journal, 52: 538-539.
LUO J, WHITE R E, ROGER B P, et al.1996.Measuring denitrification activity in soils under pasture: optimizing conditions for the short-term denitrification enzyme assay and effects of soil storage on denitrification activity [J].Soil Biology and Biochemistry, 28:409-417.
MALJANEN M, MARTIKKALA M, KOPONEN H T, et al.2007.Fluxes of nitrous oxide and nitric oxide from experimental excreta patches in boreal agricultural soil [J].Soil Biology and Biochemistry, 39:914-920.
PARSONS W F J, MITRE M E, KELLER M, et al.1993.Nitrate limitation of N2O production and denitrification from tropical pasture and rain forest soils [J].Biogeochemistry, 22: 179-193.
PHILIPS S, RABAEY K, VERSTRAETE W.2003.Impact of iron salts on activated sludge and interaction with nitrite or nitrate.Bioresource Technology, 88: 229-239.
QAFOKU N P, VAN RANST E, NOBLE A, et al.2004.Variable charge soils: their mineralogy, chemistry and management [J].Advances in Agronomy, 84:159-215.
STEHFEST E, BOUWMAN L.2006.N2O and NO emission from agricultural fields and soils under natural vegetation: summarizing available measurement data and modeling of global annual emissions[J].Nutrient Cycling in Agroecosystems, 74: 207-228.
TIEDJE J M.1982.Denitrification: Methods of Soil Analysis [M].American Society of Agronomy, USA: Madison Wisconsin:1011-1026.
XU Y B, CAI Z C, XU Z H.2012.Production and consumption of N2O during denitrification in subtropical soils of China [J].Journal of Soils and Sediments, 12:1339-1349.
XU Y B, CAI Z C.2007.Denitrification characteristics of subtropical soils in China affected by soil parent material and land use [J].European Journal of Soil Science, 58:1293-1303.
XU Y B, XU Z H, CAI Z C, et al.2013.Review of denitrification in tropical and subtropical soils of terrestrial ecosystems [J].Journal of Soils and Sediments, 13(4): 699-710.
ZHANG J, CAI Z, CHENG Y, et al.2009.Denitrification and total nitrogen gas production from forest soils of Eastern China [J].Soil Biology and Biochemistry, 41: 2551-2557.
ZHANG J, CAI Z, CHENG Y, et al.2010.Nitrate immobilization in anaerobic forest soils along a North–South transect in east China [J].Soil Science Society of America Journal, 74:1193-1200.
ZHAO W, CAI Z C, XU Z H.2007.Does ammonium-based N addition influence nitrification and acidification in humid subtropical soils of China [J]? Plant Soil, 297: 213-221.
丁昌璞.2008.中国自然土壤、旱作土壤、水稻土的氧化还原状况和特点[J].土壤学报, 45(1): 66-75.
王连峰.2003.水分前处理对土壤氧化亚氮排放的影响及其机理研究[D].南京: 中国科学院南京土壤研究所:56-58.
徐仁扣, 李九玉, 姜军.2014.可变电荷土壤中特殊化学现象及其微观机制的研究进展[J].土壤学报, 51(2): 1-9.
