在线柱切换反相高效液相色谱法测定大鼠血浆丁螺环酮浓度及药代动力学研究
2014-10-08张晓惠李晓云贾正平张娟红李文斌
张晓惠,王 荣,谢 华,尹 强,李晓云,贾正平,张娟红,李文斌
丁螺环酮是一种非苯二氮艹卓类抗焦虑药物,能够治疗高原环境下产生的焦虑、烦躁等症状,具有疗效好,不良反应少,无耐受性和依赖性等优点,缺点是首关效应明显,生物利用度低,因此,对其药代动力学的研究有助于指导临床合理用药[1],也为高原环境中应用盐酸丁螺环酮提供一定的药代动力学依据。目前对血样中丁螺环酮的检测方法主要有气相色谱-质谱法、液相色谱-质谱法、高效液相色谱法,但血样的前处理方法均为离线处理,存在操作繁琐、成本高、选择性低等缺点[2-6]。柱切换技术可实现样品的在线预处理,能克服传统离线处理方法的缺点。本实验利用实验室自制的限进性填料柱对大鼠血浆样品进行在线预处理,使丁螺环酮在预处理柱上保留并除去蛋白等大分子杂质,实现血样的直接进样分析,具有简便、快速、灵敏、准确等优点,适合生物样品中丁螺环酮的药代动力学研究。
1 仪器与试药
1.1 仪器 柱切换-反相高效液相色谱系统:2个LC-6A泵、SPD-6AV紫外检测器、CTO-6A柱温箱(日本岛津公司);7725i进样阀(美国Rheodyne公司);7000型切换阀(美国Rheodyne公司);色谱信号由SCL-6A控制器(日本岛津公司)控制;超声波清洗器(天津奥特赛恩思仪器有限公司);TGL-16B型离心机(上海安亭科学仪器厂);AE-240型电子天平(上海梅特勒-托利多)。
1.2 试药 甲醇为分析纯试剂(四川西陇化工有限公司,批号:120319),水为灭菌注射用水(西安双鹤制药有限公司,批号:110128112),丁螺环酮对照品(中国食品药品检定研究院,批号:101059-201101),丁螺环酮片(北大国际医院集团西南合成制药股份有限公司,批号:H19990302)。
2 实验方法
2.1 色谱条件 预处理柱为内表面反相限进性填料柱(45.0 mm ×4.6 mm,5.0 μm;实验室自制),预处理流动相:水 -甲醇(95∶5,V/V),流速:1 ml/min。分析柱为(Luna C18,250.0mm ×4.6 mm,5.0 μm;美国 phenomenex 公司),分析流动相:甲醇-5 mmol/L甲酸铵(75∶25,V/V),流速:1 ml/min。进样量:20 μl;柱温:25℃;检测波长:283 nm。
2.2 溶液的制备
2.2.1 标准溶液:精密称量0.01 g盐酸丁螺环酮对照品于10 ml的量瓶中,加甲醇稀释至刻度,摇匀,配成1 mg/ml的丁螺环酮储备液,置 4℃下保存。
2.2.2 内标溶液:精密称取0.001 g非洛地平标准品于10 ml的量瓶中,加甲醇稀释至刻度,摇匀,配成0.1 mg/ml的非洛地平标准溶液。
2.3 血浆样品预处理 取SPF级雄性Wistar大鼠(由中国上海斯莱克实验动物有限责任公司提供,动物合格证号:200700524909)血浆样品1 ml,置于2 ml具塞离心试管中,于5000 r/min速率下离心5 min,精密吸取上清液置于另一离心试管中,在-20℃下冷冻保存。测定样品时,在室温下融解,吸取 0.1 ml上清液,加 5 μl 0.1 mg/ml的非洛地平标准溶液。直接进样20 μl。
2.4 实验过程 进样时,切换阀如图1A连接,预处理柱与分析柱呈并联状态,因此,进样后,样品先进入预处理柱,待3 min后,血浆中的大分子蛋白等杂质随预处理流动相冲出,而丁螺环酮及其他小分子物质被保留在预处理柱上,此时,进行切换,使切换阀如图1B连接,预处理柱与分析柱呈串联状态,分析流动相将保留在预处理柱上的丁螺环酮及其他小分子物质洗脱至分析柱进行分离分析。分析完毕后,将阀切换至初始状态,使预处理流动相和分析流动相分别平衡预处理柱和分析柱5 min,以便下次进样。
3 结果
3.1 方法专属性 在上述柱切换-反相高效液相色谱条件下对空白血浆、空白血浆加对照品及大鼠血浆样品的色谱图进行比较,丁螺环酮的出峰时间为 9.6 min,内标出峰时间为 13.0 min,波长为283 nm时,丁螺环酮与内标峰完全分离,互不干扰,峰形良好,见图2。
3.2 标准曲线的绘制 取空白血浆,加丁螺环酮储备液及内标储备液,配成丁螺环酮质量浓度分别为0.2、0.3、0.6、1.2、2.4、4.8 μg/ml的系列血浆标准样品,按“2.3”项下方法处理后,精密吸取 20 μl进样,在优化的色谱条件下进行分离,测定峰面积。以峰面积与内标峰面积的比值(As/Ai)为纵坐标(Y),以丁螺环酮的质量浓度(μg/ml)为横坐标(X),绘制标准曲线,得线性回归方程为Y=0.2219 X+0.0004(r=0.9993),结果表明:血浆中的丁螺环酮在0.2~4.8 μg/ml的浓度范围内有良好的线性关系。
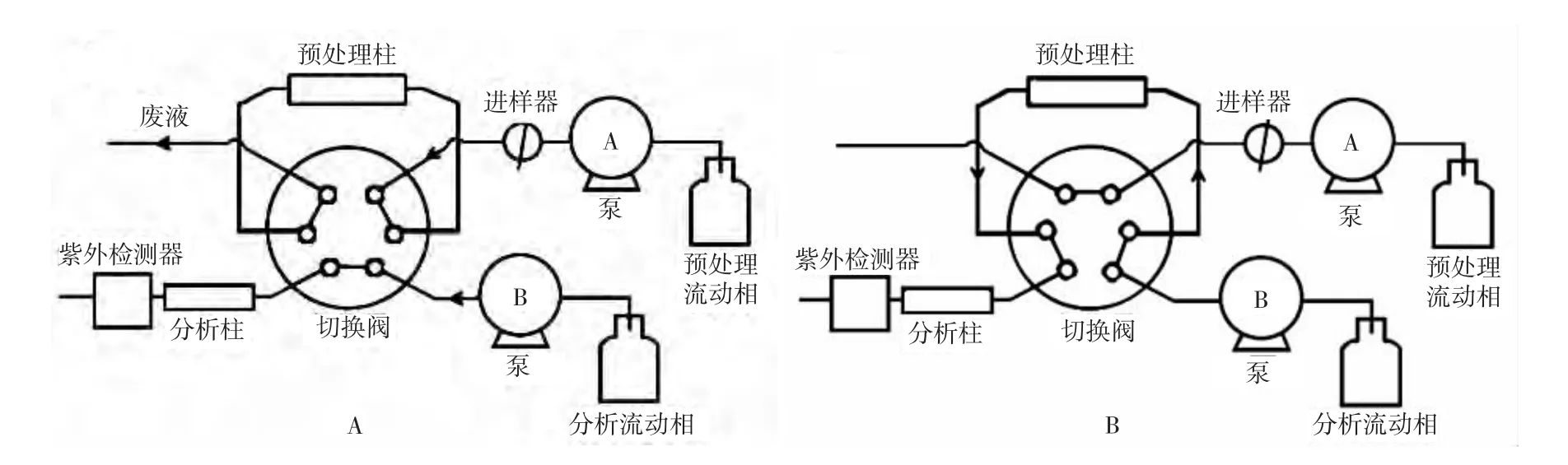
图1 柱切换-反相高效液相色谱系统示意图

图2 空白血浆、空白血浆加对照品及大鼠血浆样品高效液相色谱图
3.3 精密度及回收率 按照“3.2”项下方法配制质量浓度分别为 0.3、0.6、2.4 μg/ml的丁螺环酮标准血浆样品,按“2.3”项下方法处理后,采用柱切换-反相高效液相色谱法测定,1 d内测定6次,连续测定3 d,计算日内及日间的相对标准偏差(RSD),RSD 为 0.07%~3.54%,说明本方法精密度较好。回收率实验中,分别考查了3个加标水平0.3、0.6、2.4 μg/ml的丁螺环酮的血浆样品,处理方法同上,每个水平连续测5次,计算平均回收率,结果显示符合生物样品测定方法的要求,见表1。
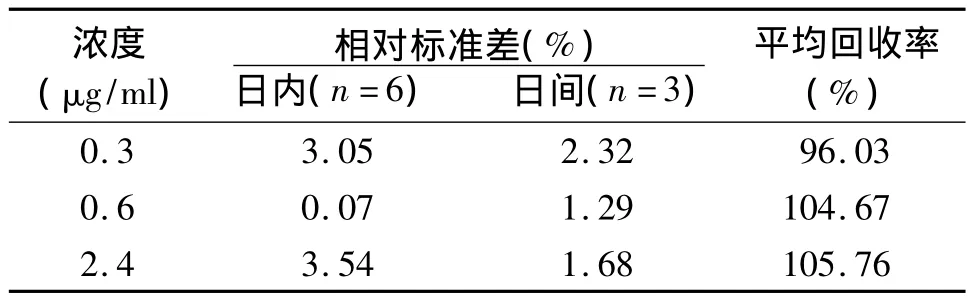
表1 3个浓度丁螺环酮的精密度和平均回收率
3.4 稳定性
3.4.1 常温放置稳定性:配制质量浓度分别为0.3、0.6、2.4 μg/ml的丁螺环酮血浆标准样品,按“2.3”项下方法处理后,于室温下放置 0、2、4、8、12、24 h,结果3个浓度的血浆标准样品RSD分别为2.00% 、1.05%、1.96% 。
3.4.2 反复冻融稳定性:配制质量浓度为0.6 μg/ml的丁螺环酮标准血浆样品,按“2.3”项下方法处理后,在 -20℃下反复冻融72 h,RSD为1.87%。
3.5 样品的测定 随机选取Wistar大鼠6只,体重200~220 g,自由饮水,禁食12 h后,按15 mg/kg剂量给予丁螺环酮片剂灌胃,并于给药后0.08、0.25、0.5、0.75、1、1.5、2、4、6、8、12、24 h 经大鼠眼球后静脉丛采血0.5 ml,置于肝素化试管中,在5000 r/min的速率下离心5 min,取上清液于-40℃冷冻保存,采用本实验建立的柱切换-反相高效液相色谱法测定大鼠体内丁螺环酮的血药浓度并绘制平均血药浓度-时间曲线(图3),采用药物动力学处理软件DAS 2.0程序对丁螺环酮的血药浓度-时间曲线进行拟合,求算药代动力学参数。其血药浓度-时间曲线面积 AUC0→8为 3.023 μg/(ml·h),AUC0→∞为4.056 μg/(ml·h),半衰期(t1/2)为3.944 h,最大浓度 (Cmax)为 1.38(μg/ml),清 除 率 (CLZ)为3.712 L/(h·kg)。

图3 丁螺环酮在大鼠体内的平均血药浓度-时间曲线
4 讨论
柱切换技术是通过改变进样阀与色谱柱、检测器之间的连接关系,通过改变流动相的走向或流动相系统,达到样品的净化、富集和分离等目的。目前已有单柱单泵、单泵双柱、双柱双泵及多柱多泵等模式,还可根据实验需要自行设计[7-8]。本文采用双泵双柱模式,由2个泵、1个进样阀、1个切换阀、限进性填料柱及分析柱组成。限进性填料是一种新的样品前处理技术,其中的内表面反相限进性填料应用较多,该填料是在硅胶表面键合一层亲水层,能够限制蛋白质的吸附,内表面具有疏水性,小分子物质可进入内孔与内表面的疏水基团相互作用而被保留,而大分子物质则不能渗透进入,因此,在死体积或接近死体积时大分子物质被洗脱下来,而不会引起蛋白在色谱柱上沉淀或不可逆吸附而造成色谱柱柱压升高及毁坏[9-13]。
限进性填料柱能够通过柱切换技术实现生物样品的在线除蛋白过程,其中切换时间是影响除蛋白的一个关键因素,切换时间过早,血浆中的蛋白还没有被完全除去,会引起蛋白在色谱柱上沉淀或不可逆吸附而造成色谱柱柱压升高或毁坏;切换过晚,会造成色谱峰展宽,分析时间延长,因此,切换时间的选择以蛋白质完全被洗脱的时间为最佳。本实验将预处理柱与检测器连接,在波长为280 nm下进空白血浆,结果表明:3 min内血浆中的蛋白质基本能被完全洗脱,因此,选择3 min为切换时间。
本文将限进性填料柱与柱切换技术相结合,建立了一种快速在线检测大鼠血浆中丁螺环酮浓度的方法,该方法可实现样品的直接进样,有效缩短了分析时间,减少血浆样品用量,降低血浆样品离线处理过程中的误差,具有较好的线性关系、精密度及回收率,操作简便,特异性好,适合生物样品中丁螺环酮的药代动力学研究,为丁螺环酮的高原药代动力学研究提供了一种新方法和依据。
[1] 朱跃华.丁螺环酮治疗广泛性焦虑症临床对照研究[J].临床精神医学杂志,2000,10(1):20-21.
[2] Foroutan S M,Zarghi A,Shafaati A R,et al.Simple high-performance liquid chromatographic determination of buspirone in human plasma[J].Farmaco,2004,59(9):739-742.
[3] Zaxariou M,Panderi I.Development and validation of a high-performance liquid chromatographicmethod for the determination of buspirone in pharmaceutical preparations[J].J Pharm Biomed Anal,2004,35(1):41-50.
[4] Gammans R E,Kerns E H,Bullen W W.Capillary gas chromatographic-mass spectrometric determination of buspirone in plasma[J].J Chromatogr,1985,345(2):285-297.
[5] Chew W M,Xu M J,Cordova C A,et al.Quantification of a cytochrome P450 3A4 substrate,buspirone,in human plasma by liquid chromatography-tandem mass spectrometry[J].J Chromatogr B Analyt Technol Biomed Life Sci,2006,844(2):235-239.
[6] 孙皎,马歆,马武翔,等.人血浆中丁螺环酮浓度的固相萃取反相高效液相色谱测定法[J].职业与健康,2006,22(12):883-884.
[7] 汪正范,杨树民,吴侔天,等.色谱联用技术[M].2版.北京:化学工业出版社,2006:258-259.
[8] Hilton J,Yokoi F,Dannals R F,et al.Column-switching HPLC for the analysis of plasma in PET imaging studies[J].Nucl Med Biol,2000,27(6):627-630.
[9] Gasparrini F,Cancelliere G,Ciogli A,et al.New chiral and restricted-access materials containing glycopeptides as selectors for the high-performance liquid chromatographic determination of chiral drugs in biological matrices[J].J Chromatogr A,2008,1191(1-2):205-213.
[10] Mullett W M.Determination of drugs in biological fluids by direct injection of samples for liquid-chromatographic analysis[J].J Biochem Biophys Methods,2007,70(2):263-273.
[11] 杨沛,王荣,马骏,等.限进填料及其在生物样品分析中的应用[J].分析仪器,2011(3):1-7.
[12] Gasparrini F,Ciogli A,D'Acquarica I,et al.Synthesis and characterization of novel internal surface reversedphase silica supports for high-performance liquid chromatography[J].J Chromatogr A,2007,1176(1-2):79-88.
[13] Sanchez A,Toledo Pinto E A,Menezes M L,et al.A simple high-performance liquid chromatography assay for on-line determination of catecholamines in adrenal gland by direct injection on an ISRP column[J].Pharmacol Res,2004,50(5):481-485.
