坏疽性脓皮病合并骨髓纤维化的研究进展
2014-10-02袁玲邹善华
袁玲 邹善华
(复旦大学附属中山医院血液科,上海 200032)
坏疽性脓皮病(pyoderma gangrenosum,PG)是一种罕见的非感染性破坏性、坏死性单发或多发皮肤溃疡,50% ~70%的PG患者合并系统性疾病,因而增加了PG诊断及治疗的难度[1]。PG患者中约半数伴发溃疡性结肠炎,关节炎、自身免疫性疾病以及血液系统疾病(如骨髓瘤、白血病、骨髓增生异常综合症等)[1]。
原发性骨髓纤维化(primary myelofibrosis,PMF)是一种慢性血液系统恶性肿瘤,与特发性血小板增多症(essential thrombocythemia,ET)和真性红细胞增多症(polycythemia vera,PV)一起归入费城染色体阴性的骨髓增殖性肿瘤[2]。PMF表现为脾肿大、骨髓病性贫血、血细胞减少、泪滴样红细胞、骨髓纤维化(myelofibrosis,MF)、髓外造血、骨髓微血管密度增加以及造血祖细胞(hematopoietic progenitor cell,HPC)和造血干细胞(hematopoietic stem cell,HSC)的结构性调整[3]。PV、ET患者经过适当的治疗后有相对较长的生存期,而PMF患者的平均生存期不超过5年。
PG大多合并血液系统疾病在内的全身性疾病,但合并MF的报道并不多。以“pyoderma gangrenosum”“myeloproliferative disorders”为 关 键 词 在PubMed检索,共检索到82篇文献(包括14篇综述),出版年为1971年至2013年;以“pyoderma gangrenosum”“fibrosis”为关键词检索,共检索到29篇文献(包括4篇综述),出版年为1974年至2013年,大部分为病例报告。国内文献检索以“骨髓纤维化”“PG”关键词在万方数据库中进行检索,无相关文献。2009年游鹏等[4]报告38例PG,其中1例合并MF。2004年渠涛等[5]报告25例 PG,其中1例合并MF。本文就PG的临床特点作一综述,并对其与MF的相关性进行初步探讨。
1 定义及流行病学特征
PG最先由 Brocq等[6]描述,而由 Brunsting等[7]在1930年命名,是一种罕见的溃疡性中性粒细胞性皮肤病,患者年龄25~54岁,发病无明显性别差异[8-9]。根据临床流行病学调查,其发病率0.3~1/100 000。在美国明尼苏达州的Mayo诊所,观察5年发现180例确诊的PG[10]。在一个对31 619例慢性腿部溃疡的调查中,PG的发生率为3%[11]。
PMF亦较为罕见,在西方国家的年发病率为0.4 ~ 1.4/100 000[12],全球发病率目前仍不清楚。PMF在白种人中更为常见,男性略多于女性,在各年龄段均可发病。诊断时的中位年龄为65岁,但仍有22%的患者发病更早,约55岁[12]。
2 病因及发病机制
2.1 PG的病因与发病机制 PG的病因与发病机制尚不明确。早年认为,PG是一种感染性疾病,但病灶早期非溃疡性皮损一般都是无菌性的,且单用抗生素无效,因而排除感染因素[7]。目前,有关PG发病机制的假说包括:(1)中性粒细胞性皮肤病。感染证据的缺乏、PG皮损中以中性粒细胞为主,证明PG的分类是一种中性粒细胞性皮肤病,PG皮肤活检显示成熟中性粒细胞浸润的炎性反应性明显特征,并已证实这些中性粒细胞伴有功能异常。在PG患者中,异常中性粒细胞转运被描述成整合素CR3和CR4表达的增加和整合素信号转导异常[13]。阻断中性粒细胞趋化和吞噬的抗中性粒细胞药物,如秋水仙碱和氨苯砜对PG有效,进一步支持了中性粒细胞功能失调在PG的发病机制方面的重要性[13];(2)越来越多的证据支持PG是一种自身免疫性疾病。PG与已知的自身免疫性疾病,如炎性反应性肠病以及关节炎、白塞病等密切相关[14-16]。PG和化脓性汗腺炎之间的关系也支持两者均属自身免疫性疾病范畴[17];(3)遗传PG以家族形式出现的报告较为罕见。近期描述的PAPA综合征(化脓性不育性关节炎、PG和痤疮)是一种影响15q常染色体的显性遗传自身免疫性疾病,PSTPIP1D基因突变可促进IL-1β细胞因子的产生以及炎性反应[18]。PG还与其他免疫遗传疾病有关,包括慢性肉芽肿性疾病、白细胞黏附缺陷以及补体 C2、C4缺陷[13];(4)细胞因子IL-8、IL-16在中性粒细胞趋化中具有重要作用,与PG的发生密切相关[19];IL-1β在PAPA综合征中的过度表达也提示这一细胞因子在PG发病中的重要作用[13]。
2.2 PMF的病因及发病机制 PMF被认为是造血干细胞克隆性增殖的结果,释放成纤维生长因子的巨核细胞和单核细胞群严重增生。PMF发病与基因突变有关,如细胞因子受体信号 Janus激酶(JAK)家族蛋白酪氨酸激酶的自体调节区段JAK2V617F失功能突变、血小板生成素受体的跨膜区段的突变(MPLW515L或MPLW515K)及其他涉及到 t(1,6)、9p、2q、3p、4 号染色体、12q 和 13q 等的基因突变[12]。然而,JAK2V617F阳性可见于90%的PV患者,而约50%的PMF患者并无JAK2或cMPL突变,因此基因突变并非PMF的唯一原因[12]。部分 PMF患者有细胞因子、抗体、补体异常,且激素治疗有效,提示该类患者存在免疫异常。细胞因子 IL-6、IL-8、TNFα、IL-2、IFN-γ 的表达可能与临床症状,如MF、血管生成、全身症状和恶病质相关[20]。免疫调节药物抗细胞因子和抗血管生成剂,在改善MF的血细胞减少(尤其是贫血和血小板减少症)以及缩小肿大脾脏方面效果较好[12,21]。
2.3 PG与PMF共同的发病机制 PMF与PG的共患病提示两者之间可能涉及共同的发病机制,有关其共发病机制的假说包括以下方面。
2.3.1 细胞因子 细胞因子在PMF与PG的发病中均有关键作用。TGF-β、TNF-α、IL-1、IL-8 等细胞因子均可能参与发病过程。IL-8是一种重要的中性粒细胞趋化因子,在PG中过度表达;重组IL-8转染人皮肤异种移植细胞可诱导PG样溃疡[19];而IL-8在HPC的增殖、动员和血管新生等方面具有多种生物学活性,IL-8在PMF患者血清中也呈高表达。TGF-β是成纤维细胞分裂因子,对MF的发生起决定性作用[12];TGF-β1 敲除的小鼠不会发生 MF[12]。IL-1对于单核细胞产生TGF-β是必需的,其与神经激肽κB(NF-κB)的协同作用共同导致了纤维化;PAPA综合征中应用IL-1抑制剂阿那白滞素可改善患者PG病变,提示IL-1在PG的发病机制中具有重要作用[13]。
2.3.2 免疫异常 PG可与单克隆丙种球蛋白病合并存在,而在部分PMF中也可检测到自身抗体,如抗核抗体、抗心磷脂抗体、抗基质蛋白抗体等。此外,Coombs试验呈阳性反应;循环免疫复合物、补体活性及免疫球蛋白增高和或免疫球蛋白IgG、IgA、IgM异常抗体的出现,均提示体液免疫可能也参与PG和PMF共同发病[22]。在细胞免疫方面,两者也可能存在共性,抗原刺激的T细胞攻击皮肤引发PG[13];糖皮质激素可能是通过抑制过量的细胞毒T淋巴细胞而在PMF治疗中起效[12],虽然T淋巴细胞计数和吞噬功能正常,但在体外实验中却无白细胞迁移,提示中性粒细胞的内在异常可能是诱发此病的主要原因[23]。免疫治疗,如糖皮质激素、沙利度胺等治疗的有效性也进一步验证了两者具有共同的免疫异常的假设[12-13]。
3 临床表现
PG典型的皮损开始于小结节、斑块或无菌性脓疱,数天后扩大并侵蚀成为一个边界清晰的溃疡,伴潜行的紫罗兰色的边界以及周围区域的红斑(图1),疼痛明显[24]。皮肤和周围出现坏死,形成了易碎的伤口床,经常会有出血性或脓性渗出物,有时延伸至肌肉[1]。皮损愈合后呈现Cribiform或“筛状”萎缩性瘢痕(图2)。PG皮损具有多形性及复发性[13],25% ~50%PG患者的皮损发生于创伤部位,被称为超敏反应[8]。成人PG的皮损最常累及下肢,任何解剖部位均可受累[8,13];而在儿童(大约占4%的病例),典型皮损累及下肢、臀部、会阴区以及头部和颈部[13]。PG也可以影响皮肤外部位,如眼部(巩膜炎和眼眶炎性反应)、肺部(无菌结节),脾脏和肌肉骨骼(表现为无菌化脓性关节炎和中性粒细胞肌炎)[13]。临床上根据皮损形态将PG分为溃疡型、脓疱型、大疱型、增殖型及造口周围型,其中以溃疡型最常见[13]。
约50%的PG患者有潜在的全身疾病,甚至有报道其相关性可达78%[2,8];其余PG病例被认为是特发性病例。炎性反应性肠病、关节炎和血液系统疾病是最为常见的PG相关疾病。大部分PG病例是在全身疾病诊断后确诊,但也存在潜在疾病先兆的病例[8,25]。
为了更充分描述PG相关疾病谱,Ahronowitz I等以“pyoderma gangrenosum”为关键词在PubMed中进行检索。此检索除外了单一病例报告,最后的数据规定限于病例系列,详尽讨论了最常见的合并疾病,也枚举了相对少见但可能发生的少见疾病如化脓性汗腺炎、系统性红斑狼疮、HIV等,并据此改写了PG 的分类方法[13]。

图1 1例骨髓纤维化合并坏疽性脓皮病女性患者的皮损
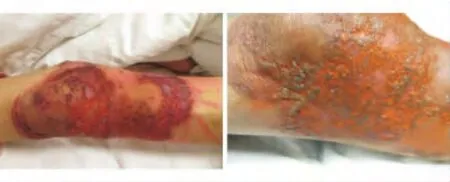
图2 图1同例患者右膝部病灶愈合后显示“筛状”萎缩性疤痕
3.1 炎症性肠病(inflammatory bowel disease,IBDs)IBD,如克隆恩病和溃疡性结肠炎(uicerative colitis,UC)是最常报告的与PG相关的系统性疾病。Ahronowitz等[13]对214例PG病例进行回顾分析发现,IBD与PG的相关性达41%。PG合并UC的患者进行全直肠切除术后PG得以缓解,这支持了两者之间的相关性[8]。一项对2402例法国IBD患者的前瞻性研究中,PG仅发生于0.75%的患者,与普通人群相似。迄今有关PG最大样本量样的报告来自于德国,该研究选取20个皮肤伤口护理中心的259例患者,其中仅9.3%的患者合并慢性炎症性肠病或类风湿关节炎[15];该研究和以往研究结果的差异原因仍不明确,但提示既往可能夸大了两者之间的关联性[26]。
3.2 关节炎 PG常伴发各种关节炎,最常见的是累及单个、大关节的血清阴性的关节炎[27]。有报告[13]显示,两者相关性甚至达37%。关节炎的临床严重性与PG活动度无关。
3.3 血液系统异常、异常蛋白血症与恶性肿瘤 血液系统疾病及其他肿瘤均为PG常见的合并症。Bolognia等[28]的研究提示,PG合并恶性肿瘤的发生率高达25%。2011年Binus等[15]对103例PG的回顾研究发现,21例(20%)合并血液系统异常;而Ahronowitz等[13]发现,214例 PG 中,血液系统恶性肿瘤或其他血液系统疾病(除单克隆丙种球蛋白病外)有35例(16%),另有36例(17%)单克隆丙种球蛋白病。Al Ghazal等[16]对259例PG的分析提示,恶性肿瘤占12.4%,其中有血液系统肿瘤史者占 3.9%。
血液系统恶性肿瘤或其他血液系统疾病(除单克隆丙种球蛋白病外)涉及11篇文献,共35名患者,5例急性白血病,8例慢性白血病,3例淋巴瘤,3例真性红细胞增多症,2例骨髓瘤,10例骨髓增生异常综合症,2例MF/髓样化生,2例其他血液系统疾病[13]。虽然单克隆丙种球蛋白病患者可能呈临床良性过程,但部分患者也会进展为骨髓瘤。有关单克隆丙种球蛋白血症进展是否与PG相关还未见相关报道。
典型的溃疡性PG是最常见的合并单克隆丙种球蛋白血症的亚型。血液系统恶性肿瘤见于7%的PG病例,急性髓性白血病(acute myeloid leukemia,AML)最常见。
PG皮损的发展预示着AML预后较差,合并PG的AML患者1年病死率达75%。临床观察提示,对大疱型PG皮损患者有必要进行迅速、彻底的检查。同样,在骨髓增生异常综合症和骨髓增殖性疾病中,PG的进展也可能提示恶性转化。在潜在恶性肿瘤得到治疗后,大疱型PG会得以改善。在部分白血病伴发大疱型PG患者的皮损中可见白血病细胞浸润[13]。
PG合并MF的报告相对较少。Gopinath等[29]于1974年首次报告两者相关,在25篇文献报道中几乎均为单病例报告。然而,合并MF的PG患者,PG常发生于少见部位。Young等[30]报告了1例女性患者行脾切除后PG发生于手部;2007年Zivanovic等[31]报告1例MF患者被诊断出发生于手背侧的典型PG皮损,而PG的非典型形式可能是恶性肿瘤的第一迹象,尤其是骨髓增生性疾病患者。
4 组织病理检查
尽管活检本身可能会诱发PG的超敏反应,但皮肤活检仍然十分重要,因其有助于排除其他原因引起的溃疡,尤其是感染[16,32]。为了获取较理想的标本,应尽可能作一个较深的椭圆切口活检,标本应包括病灶边缘的一部分和皮下组织[33]。可针对细菌感染用苏木精或特殊染色作组织学检查,同时进行细菌、病毒、真菌和非典型分枝杆菌培养[13]。直接免疫荧光检查显示,大部分PG活检标本均有IgM、C3和纤维蛋白沉积在真皮乳头层和网状层的血管上。活检可能对排除自身免疫性皮肤炎或血管炎有帮助(虽然这些指标对PG并无敏感性和特异性)[33]。早于溃疡出现的最初病变显示一个深部化脓性病灶,常常是有密集中性粒细胞浸润的滤泡中心性炎性反应,且白细胞血管炎也经常出现。不能确定的溃疡边界显示混合性、中性粒细胞为主的炎性浸润,很典型的病变的基底会显示坏死和出血的证据[33]。有1例来自于PG溃疡周围红斑的标本,可见到皮肤或脂肪血管的血栓形成的坏死,并伴有淋巴细胞为主的浸润[34]。PG病灶也可见水肿和淋巴细胞血管炎。
5 实验室检查
有关PG诊断的实验室检查并无特异性,但应作为排除感染或恶性肿瘤的初步诊断,尤其是在患者接受全身系统性免疫抑制治疗前,适当的实验室检查必不可少。常规实验室检查,如全血细胞计数及分类、电解质、尿常规和肝功能可能有助于初始诊断血液系统疾病与肝或肾功能衰竭以及肝炎。其他检查,如抗核抗体、抗磷脂抗体、冷球蛋白、类风湿因子、循环抗中性粒细胞胞浆抗体等,对于排除系统性疾病有一定帮助。进一步对伴随症状的检查手段包括胸部X线片(感染或系统性血管炎)、IBD患者行粪隐血和乙状结肠镜或结肠镜检查,血液病相关检测(血清蛋白电泳,随机尿蛋白或尿蛋白电泳,免疫固定电泳,外周血涂片以及骨髓活检)。感染病因检查包括HIV检测、乙型肝炎血清学检查和快速血浆反应素试验[13]。
6 诊断与鉴别诊断
PG作为临床诊断,缺乏特异血清学和组织学指标,目前普遍采用Su等[34]在2004年提出的标准,需满足两个主要标准:(1)疼痛、坏死、不规则、紫色、边界被破坏的皮肤溃疡的快速进展(每天扩大1~2 cm,或1个月内溃疡增加超过50%)。(2)其他原因所致的皮肤溃疡除外,并满足以下至少两条,包括:既往史有超敏反应性或临床发现筛状瘢痕;PG相关的系统性疾病;组织病理学检查(无菌性嗜中性粒细胞皮肤病±混合性炎性反应±淋巴细胞性脉管炎);治疗反应(对类固醇激素系统性治疗快速有效)。
PG常被误诊为其他疾病。PG诊断必须严格排除其他病因所致的溃疡,尤其是感染因素。鉴别诊断的范围很广,需与PG鉴别的疾病包括:(1)血管闭塞或淤积如抗磷脂抗体综合征、静脉淤积性溃疡、Klippel-Trenauney-Weber综合征、小血管闭塞性动脉病等;(2)血管炎,如Wegener肉芽肿病、结节性多动脉炎、大动脉炎等;(3)恶性疾病的皮肤受累如T细胞淋巴瘤、大疱性蕈样肉芽肿、皮肤白血病、朗格汉斯细胞性组织细胞增生症;(4)其他副肿瘤性皮肤病,如Sweet综合症、皮肌炎、副肿瘤性天疱疮、坏死松解性游走性红斑等;(5)原发性皮肤感染,包括孢子丝菌病、曲菌病、隐球菌病、Ⅱ型单纯疱疹、皮肤结核病、皮肤阿米巴病、接合菌病、马尔尼菲青霉菌病;(6)药物诱发性和外因性溃疡,如羟基脲诱发的溃疡、溴疹、接触性女阴炎、药源性狼疮、滥用注射药物伴继发感染;(7)其他炎症性疾病:皮肤Crohn病、溃疡性类脂质渐进性坏死[35]。
7 治 疗
7.1 PG的治疗 PG的治疗尚无统一的规范。PG既有全身炎性反应参与,又有伤口,有效的治疗策略必须同时兼顾这两方面[36]。治疗的目的是控制炎性反应,减少疼痛,优化创面愈合并减少恶化因素。治疗需要根据皮损大小、数量、位置、类型、病变形成过程以及潜在疾病决定[13],主要的治疗包括局部治疗和全身治疗。最初的治疗选择应开始于优化伤口护理,这往往是PG治疗成功的关键,有效的伤口护理对于减轻疼痛、减少继发感染率有较大作用;联合NSAIDs、阿片类等药物以控制疼痛[37];局部治疗,还包括局部应用抗生素、抗中性粒细胞药物、糖皮质激素及其他免疫抑制剂[38-39],尤其是对于需要排除隐匿性感染或恶性肿瘤的患者。但仍需注意的是,虽然外用药比全身治疗药物的不良反应和禁忌症更少,但需要频繁使用,且仅限于轻症及表面病变的处理,因而对于PG快速进展的患者,全身免疫抑制治疗更为重要。目前针对PG最常用的治疗建议是:一经诊断,首先选择糖皮质激素全身治疗,即强的松0.5 ~ 1 mg/(kg·d),甲基强的松龙 0.8 mg/(kg·d)[40-41],可阻止病变进展[37]。生物治疗药物英夫利昔单抗是抗TNFα单克隆抗体,它是唯一一个在PG治疗中有疗效的生物药物(I级证据)[42]。秋水仙碱、磺胺类和砜类等抗中性粒细胞治疗药物通过影响中性粒细胞趋化而在PG的治疗中起效[13]。环孢霉素(口服 3 ~10mg/(kg·d)[43]或静脉滴注4 mg/(kg·d),7~22 d后序贯口服4~7 mg/(kg·d)[44])已被证实对 PG 有效,目前认为环孢霉素也可作为PG的一线治疗,尤其是对病情进展快的患者。其他免疫治疗如沙利度胺联用或不联用糖皮质激素[45-47]以及静脉注射免疫球蛋白[48]也在部分PG患者中有效。
7.2 骨髓纤维化的治疗 目前PMF、PV后MF或ET后MF的治疗大多为姑息治疗,常用的药物并不直接针对疾病本身潜在的细胞学或遗传学的病变[49]。自体干细胞移植(ASCT)是唯一可能治愈MF的治疗方式,尤对那些有高危转白倾向的患者应早期选择[49-50]。MF最常见的症状是严重贫血和/或脾肿大及血小板减少[49],输血及应用达那唑、促红细胞生成素、羟基脲或低剂量美法仑[49-50]、低剂量沙利度胺/强的松[12,21,51-52]、雷利度胺[52-54]可能有助于改善上述症状。酪氨酸激酶抑制剂、TGF-β抑制剂、NFκB抑制剂、地西他滨、Bcl-xL抑制剂以及蛋白酶抑制剂等的临床试验也正在进行[4]。
7.3 PG与PMF的治疗 关于PMF与PG共同发病报道较少,因而无确切的治疗方案。基于两者均有免疫因素参与发病,可选择协同免疫治疗的药物,沙利度胺及其衍生物联合糖皮质激素有望成为新的的治疗选择。沙利度胺除了对治疗多发性骨髓瘤具有良好的疗效外,其在皮肤病治疗方面也得到了较为广泛的应用,1998年FDA批准其用于控制急性麻风的皮肤症状。沙利度胺是一种免疫调节药物,可通过多种免疫机制起作用;可影响肿瘤坏死因子-α(TNF-α)、白细胞介素-2(IL-2)、IL-6、IL-12、IL16-21和IFN-γ等多种细胞因子,并协同糖皮质激素抑制细胞因子的作用;同时,其在体液免疫及细胞免疫方面也起作用,可通过抑制中性粒细胞的趋化性,发挥对皮肤病的治疗作用。沙利度胺也是PG的治疗选择[55]。有研究对79例伴髓样化生的MF患者给予100~800 mg/d的口服沙利度胺治疗,结果显示,患者贫血(0~50%)、血小板减少(25% ~80%)均有明显改善,但对脾肿大改善不明显。然而,该治疗剂量的沙利度胺很难被耐受,91%的髓样化生的骨髓纤维化(MMM)患者因为其不良反应而中止了治疗;并且在治疗过程中部分MF患者出现了骨髓过度增殖[56]。Mesa等[21]对21 例 PMF 患者给予 3 个月的小剂量口服沙利度胺(50 mg/d)和逐渐减量[0.5 mg/(kg·d)为第1 个月;0.25 mg/(kg·d)为第2个月;0.125 mg/(kg·d)为第3个月]的口服强的松(THAL-PRED方案),患者的贫血、血小板减少有明显改善。上述研究结果证实,小剂量沙利度胺联合糖皮质激素方案对于改善MF患者的贫血及血小板减少等方面症状有效,并且耐受良好。有关沙利度胺衍生物来那度胺的研究更多集中于肿瘤性疾病。在PMF的治疗中,Holle等[57]对7例沙利度胺治疗无反应的PMF患者应用来那度胺后,其中4例临床起效。来那度胺联合泼尼松似乎比单药沙利度胺或来那度胺更有效和安全[58]。Quintas-Cardama等[59]对40例MF患者给予口服来那度胺联合泼尼松治疗,在改善贫血及脾肿大方面的反应率分别为30%和42%;10/11例初始治疗MF 4级的患者出现网状纤维减少至少2分;所有8例JAK2阳性患者均出现基线等位基因负荷的减低,其中1例的突变已检测不到。因此,对于MF的治疗,来那度胺联合泼尼松会诱导更为持久的临床、分子及病理反应。
8 预 后
有关PMF的预后,目前最常用的是国际动态预后评分系统加强版(DIPSS-plus),包括年龄、全身症状、血红蛋白、血小板数、白细胞数、外周血原始细胞、是否输血依赖以及不良/复杂核型8个因素,其低危、中危-1、中危-2及高危组的中位生存期分别为15.4、6.5、2.9 及 1.3 年[12]。
研究[9]提示,与特发性PG相比,全身性疾病合并PG患者具有更高的复发率和较差的临床结果。对42例患者平均随访26.5个月的研究[60]显示,特发性组和疾病相关组PG预后并无显著差异;总复发率为56%。
Bennett等[35]对86例 PG患者的回顾分析发现,经典型PG平均缓解时间为(11.5±11.1)个月,而大疱性仅(9 ±13.7)个月(P=0.03),其中有5 例PG经多种治疗仍复发。
9 结 语
PG和MF发病率均较低,两种疾病同时发病更为罕见。然而两者在发病机理上的共性,尤其是细胞因子等免疫因素在发病中的重要作用,为这两种疾病的治疗带来了契机,沙利度胺联合泼尼松似乎是目前疗效较好且经济实用的方案。而沙利度胺衍生物因其在MF治疗方面优于沙利度胺的疗效,使其对于共患此两类疾病的患者有更大的临床应用前景。
[1]Crowson AN,Mihm MC Jr,Magro C.Pyoderma gangrenosum:a review[J].J Cutan Pathol,2003,30(2):97-107.
[2]Tefferi A,Vardiman JW.Classification and diagnosis of myeloproliferative neoplasms:The 2008 World Health Organization criteria and point-of-care diagnostic algorithms[J].Leukemia,2008,22:14-22.
[3]Hoffman R,Rondelli D.Biology and treatment of primary myelofibrosis[J].Hematology Am Soc Hematol Educ Program,2007,346-354.
[4]游鹏,何晓玲.38例坏疽性脓皮病的临床分型与治疗的临床研究[J].中国现代医生,2009,47(14):51-52.
[5]渠涛,王宝玺.坏疽性脓皮病25例回顾性分析[J].中国皮肤性病学杂志,2004,18(10):598-600.
[6]Brocq L.Nouvelle contribution a l’etude du phagedenisme geometrique[J].Ann Dermatol Syphil,1916,6(1):1-39.
[7]Brunsting LA,Goeckerman WH,O'Leary PA.Pyoderma gangenosum:clinical and experimental observations in five cases occurring in adults[J].Arch Dermatol,1930,22:655.
[8]Powell FC,Schroeter AL,Su WP,et al.Pyoderma gangrenosum:a review of 86 patients[J].Q J Med,1985,55(217):173-186.
[9]Powell FC,Su WP,Perry HO.Pyoderma gangrenosum:classification and management[J].J Am Acad Dermatol,1996,34(3):395-409.
[10]Powell FC,Schroeter AL,Su WP,et al.Pyoderma gangrenosum and sarcoidosis[J].Arch Dermatol,1984,120(7):959-960.
[11]Körber A,Klode J,Al-Benna S,et al.Etiology of chronic leg ulcers in 31,619 patients in Germany analyzed by an expert survey[J].J Dtsch Dermatol Ges,2011,9(2):116-121.
[12]Tabarroki A,Tiu RV.Immunomodulatory agents in myelofibrosis[J].Expert Opin Investig Drugs,2012,21(8):1141-1154.
[13]Ahronowitz I,Harp J,Shinkai K.Etiology and management of pyoderma gangrenosum:a comprehensive review[J].Am J Clin Dermatol,2012,13(3):191-211.
[14]Pereira N,Brites MM,Goncalo M.Pyoderma gangrenosum--a review of 24 cases observed over 10 years.Int J Dermatol,2013,52(8):938-45
[15]Binus AM,Qureshi AA,Li VW.Pyoderma gangrenosum:a retrospective review of patient characteristics,comorbidities and therapy in 103 patients[J].Br J Dermatol,2011,165(6):1244-1250.
[16]Al Ghazal P,Herberger K,Schaller J,et al.Associated factors and comorbidities in patients with pyoderma gangrenosum in Germany:a retrospective multicentric analysis in 259 patients[J].Orphanet J Rare Dis,2013,8(1):136.
[17]Braun-Falco M,Kovnerystyy O,Lohse P,et al.Pyoderma gangrenosum,acne,and suppurative hidradenitis(PASH):a new autoinflammatory syndrome distinct from PAPA syndrome[J].J Am Acad Dermatol,2012,,6(3):409-415.
[18]Shoham NG,Centola M,Mansfield E,et al.Pyrin binds the PSTPIP1/CD2BP1 protein,defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway[J].Proc Natl Acad Sci U S A,2003,100(23):13501-13506.
[19]Oka M,Berking C,Nesbit M,et al.Interleukin-8 overexpression is present in pyoderma gangrenosum ulcers and leads to ulcer formation in human skin xenografts[J].Lab Invest,2000,80(4):595-604.
[20]Lacy MQ,Tefferi A.Pomalidomide therapy for multiple myeloma and myelofibrosis:an update[J].Leuk Lymphoma,2011,52:560-566.
[21]Mesa RA,Steensma DP,Pardanani A,et al.A phase 2 trial of combination low-dose thalidomide and prednisone for the treatment of myelofibrosis with myeloid metaplasia[J].Blood,2003,101(7):2534-2541.
[22]任悦,付蓉,邵宗鸿.特发性骨髓纤维化发病机制的研究进展[J].中华血液学杂志,2008,29(11):787-789.
[23]Deschamps P,Leroy D,Dresch C,et al.Immune deficiences during pyoderma gangrenosum associated with a polycythemia vera[J].Nouv Presse Med,1977,25,6(26):2339-2341.
[24]Su WPD,Davis MDP,Weenig RH,et al.Pyoderma gangrenosum:clinicopathologic correlation and proposed diagnostic criteria[J].Int J Dermatol,2004,43(11):790-800.
[25]Ruocco E,Sangiuliano S,Gravina AG,et al.Pyoderma gangrenosum:an updated review[J].J Eur Acad Dermatol Venereol,2009,23(9):1008-1017.
[26]Farhi D,Cosnes J,Zizi N,et al.Significance of erythema nodosum and pyoderma gangrenosum in inflammatory bowel diseases:a cohort study of 2402 patients[J].Medicine(Baltimore),2008,87(5):281-293.
[27]Charles CA,Leon A,Banta MR,et al.Etanercept for the treatment of refractory pyoderma gangrenosum:a brief series[J].Int J Dermatol,2007,46(10):1095-1099.
[28]Bolognia J,Jorizzo J,Rapini R:Pyoderma gangrenosum[J].Dermatology,2003,1:415-418.
[29]Gopinath DK,Wolfe RD,Sabharwal K,et al.Pyoderma gangrenosum with myelofibrosis[J].J Ky Med Assoc,1974,72:549-550,566.
[30]Yang VL,Fernando B,Tabas M,et al.A case study of pyoderma gangrenosum.J Hand Surg AM,1988,13(2):259-262.
[31]Zivanovic D,Tanasilovic S,Skiljevic D,et al.Atypical pyoderma gangrenosum in a patient with osteomyelofibrosis[J].Vojnosanit Pregl,2007,64(11):787-789.
[32]Barr KL,Chhatwal HK,Wesson SK,et al.Pyoderma gangrenosum masquerading as necrotizing fasciitis.Am J Otolaryngol 2009 Aug,30(4):273-276
[33]Su WP,Schroeter AL,Perry HO,et al.Histopathologic and immunopathologic study of pyoderma gangrenosum[J].J Cutan Pathol,1986,13(5):323-330.
[34]Su WPD,Davis MDP,Weenig RH,et al.Pyoderma gangrenosum:clinicopathologic correlation and proposed diagnostic criteria[J].Int J Dermatol,2004,43(11):790-800.
[35]顾有守.坏疽性脓皮病[J].临床皮肤科杂志,2005,34(10):708-709.
[36]Bennett ML,Jackson JM,Jorizzo JL,et al.Pyoderma gangrenosum:a comparison of typical and atypical forms with an emphasis on time to remission.Case review of 86 patients from 2 institutions[J].Medicine(Baltimore),2000,79(1):37-46.
[37]Miller J,Yentzer BA,Clark A,et al.Pyoderma gangrenosum:a review and update on new therapies[J].J Am Acad Dermatol,2010,62(4):646-654.
[38]Wenzel J,Gerdsen R,Phillipp-Dormston W,et al.Topical treatment of pyoderma gangraenosum[J].Dermatology(Basel),2002,205(3):221-223.
[39]Le Cleach L,Moguelet P,Perrin P,et al.Is topical monotherapy effective for localized pyoderma gangrenosum?[J].Arch Dermatol,2011,147(1):101-103.
[40]Gettler S,Rothe M,Grin C,et al.Optimal treatment of pyoderma gangrenosum[J].Am J Clin Dermatol,2003,4(9):597-608.
[41]Reichrath J,Bens G,Bonowitz A,et al.Treatment recommendations for pyoderma gangrenosum:an evidence-based review of the literature based onmore than 350 patients[J].J Am Acad Dermatol,2005,53(2):273-283.
[42]Brooklyn TN,Dunnill MGS,Shetty A,et al.Infliximab for the treatment of pyoderma gangrenosum:a randomised,double blind,placebo controlled trial[J].Gut,2006,55(4):505-509.
[43]Elgart G,Stover P,Larson K,et al.Treatment of pyoderma gangrenosum with cyclosporine:results in seven patients[J].J Am Acad Dermatol,1991,24(1):83-86.
[44]Friedman S,Marion JF,Scherl E,et al.Intravenous cyclosporine in refractory pyoderma gangrenosum complicating inflammatory bowel disease[J].Inflamm Bowel Dis,2001,7(1):1-7.
[45]Paghdal KV,Schwartz R.Thalidomide and its dermatologic uses[J].Acta Dermatovenerol Croat,2007,15(1):39-44.
[46]Chen M,Doherty SD,Hsu S.Innovative uses of thalidomide[J].Dermatol Clin,2010,28(3):577-586.
[47]Wu JJ,Huang DB,Pang KR,et al.Thalidomide:dermatological indications,mechanisms of action and side-effects[J].Br J Dermatol,2005,153(2):254-273.
[48]de Zwaan SE,Iland HJ,Damian DL.Treatment of refractory pyoderma gangrenosum with intravenous immunoglobulin[J].Australas J Dermatol,2009,50(1):56-59.
[49]Hoffman R,Ravandi-Kashandi F.Idiopathic myelofibrosis.In:Hoffman R,Benz EJ Jr.Shattil SJ,et al.Eds.Hematology:Basic Principles and Practices,4th edition[J].Philadelphia,PA:Elsevier Scientific,2005:1255-1257.
[50]Mesa RA,Li CY,Ketterling RP,et al.Leukemic transformation in myelofibrosis with myeloid metaplasia:a single institution experience with 91 cases[J].Blood,2005,105:973-977.
[51]Thapaliya P,Tefferi A,Pardanani A,et al.International working group for myelofibrosis research and treatment response assessment and long-term follow-up of 50 myelofibrosis patients treated with thalidomide-prednisone based regimens[J].Am J Hematol,2011,86(1):96-98.
[52]Holle N,de Witte T,Mandigers C,et al.Thalidomide and lenalidomide in primary myelofibrosis[J].Neth J Med,2010,68(1):293-298.
[53]Quintas-Cardama A,Kantarjian HM,Manshouri T,et al.Lenalidomide plus prednisone results in durable clinical,histopathologic,and molecular responses in patients with myelofibrosis[J].J Clin Oncol,2009,27(28):4760-4766.
[54]Mesa RA,Yao X,Cripe LD,et al.Lenalidomide and prednisone for myelofibrosis:Eastern Cooperative Oncology Group(ECOG)phase 2 trial E4903[J].Blood,2010,116(22):4436-4438.
[55]Peuckmann V,Fisch M,Bruera E.Potential novel uses of thalidomide:focus on palliative care[J].Drugs,2000,60(2):273-292.
[56]Barosi G,Grossi A,Comotti B,et al.Safety and efficacy of thalidomide in patients with myelofibrosis with myeloid metaplasia[J].Br J Haematol,2001,114(1):78-83.
[57]Holle N,de Witte T,Mandigers C.Thalidomide and lenalidomide in primary myelofibrosis[J].Neth J Med,2010,68(1):293-298.
[58]Jabbour E,Thomas D,Kantarjian H,et al.Comparison of thalidomide and lenalidomide as therapy for myel ofibrosis[J].Blood,2011,118(4):899-902.
[59]Quintas-Cardama A,Kantarjian HM,Manshouri T,et al.Lenalidomide plus prednisone results in durable clinical,histopathologic,and molecular responses in patients with myelofibrosis P[J].J Clin Oncol,2009,27(28):4760-4766.
[60]von den Driesch P.Pyoderma gangrenosum:a report of 44 cases with followup[J].Br J Dermatol,1997,137(6):1000-1005.
