C原子在Cu(200)表面吸附的第一原理研究
2014-09-21付志雄毕冬梅赵利军
付志雄,毕冬梅,赵利军
(长春大学 理学院,长春 130022)
0 引言
2004年,英国曼切斯特大学的科学家利用胶带剥离高定向热解石墨获得了独立存在的高质量石墨烯[1]。石墨烯是由单层C原子紧密堆积成的二维蜂窝状结构,是构成其他维数碳材料的基本结构单元[2]。石墨烯具有优异的电学特性、良好的透光性和机械特性[3]。在超级电容器、锂离子电池以及燃料电池等方面具有广泛的应用前景[4]。
化学气相沉积方法是制备大面积高质量石墨烯的主要方法,常用的性能最好的催化剂是Cu和Ni。2009年,研究人员首次在Cu衬底表面生长出单层占95%的石墨烯[5]。C在Cu中的固溶度很低,在Cu衬底上石墨烯的生长机制为表面生长,即高温下气态碳源裂解生成的C原子吸附在金属表面,进而成核生长成“石墨烯岛”,并通过“石墨烯岛”的二维长大合并得到连续的石墨烯[6]。因此,C原子在Cu表面的吸附是Cu衬底上化学气相沉积制备石墨烯的重要步骤。第一原理计算已被广泛用于研究原子或分子在表面的吸附,例如,研究人员对C原子在Ni表面和亚表面的吸附做了详细分析,通过研究C原子的扩散来说明Ni表面碳纳米管和碳纳米纤维的生长[7]。
本文采用第一原理密度泛函理论研究C原子在Cu(200)表面的吸附,计算C原子在Cu(200)表面的吸附能,探讨C原子吸附对Cu(200)表面的几何结构、电荷分布和差分电荷密度的影响,为揭示Cu表面石墨烯的生长机理提供理论指导。
1 计算方法
本文的所有计算结果均由基于第一原理密度泛函理论的CASTEP软件[8]得到。在这个软件包中进行非自旋极化的结构优化、总能量和性质计算,电子间的交换关联泛函采用基于广义梯度近似的Perdew-Burke-Ernzerhof[9]函数,采用超软赝势[10]描述离子实和价电子之间的相互作用,平面波的截断能为400 eV。结构优化采用BFGS算法,单原子能量收敛标准为1.0×10-5eV,原子间相互作用力收敛标准为0.03 eV,应力收敛标准为0.05 GPa,原子最大位移收敛标准为1.0×10-3Å。
采用超晶胞模型来模拟Cu(200)表面,选用2×2的表面,Cu原子层取6层,每一层4个原子,相邻表面层之间有15Å的真空层。C原子吸附在Cu(200)表面上,覆盖率为0.25%。计算中最下面3个Cu原子层固定,表面的3个Cu原子层和吸附的C原子进行驰豫,直到达到体系的总能量最小,得到最稳定的表面吸附结构。
2 结果与讨论
Cu晶体属于面心立方结构,如图1(a)所示,晶格常数经过优化后为3.62Å,Cu晶体中Cu-Cu键键长为2.56Å。Cu(200)表面是较为规则的低指数面,经过结构弛豫,并没有发现有表面重构,只是表面几层原子在垂直于表面的方向发生了移动。对于表面C原子吸附,如图1(b)所示,Cu(200)表面共有3个高对称吸附位置,分别是四重空位(Hollow位)、二重空位(Bridge位)和一重顶位(Top位)。C原子在Top位的吸附是不稳定的,几何优化后Top位的C原子会自动扩散到邻近的Hollow位。

图1 Cu晶体的结构和C原子在Cu(200)表面的三种高对称吸附位置
C原子在Cu(200)表面吸附的结构参数如表1所示。当C原子处于表面Hollow位和Bridge位时,吸附的C原子与表面的Co原子分别呈四配位和两配位成键。当C原子处于Hollow位和Bridge位时,吸附位置附近的Cu-Cu键的键长分别为2.65Å和2.69Å,比Cu晶体中Cu-Cu键的键长稍长,这说明C原子在表面的吸附使表面的Cu-Cu键发生扩张。当C原子处于Hollow位时,Cu-C键键长为1.90Å,比C原子处于Bridge位时的Cu-C键键长要长,这是因为在Hollow位时,C原子的配位数较大。当C原子处于Hollow位和Bridge位时,C原子到Cu表面的垂直距离分别为0.29Å和1.20Å,在Hollow位时,C原子不仅与表面的四个Cu原子成键,还可以与其下方第二层的Cu原子有相互作用。
表1C原子在Cu(200)表面Hollow位和Bridge位吸附时的结构参数,其中EA表示吸附能,dCu-Cu表示吸附位置附近Cu-Cu键平均键长,dCu-C表示Cu-C键平均键长,h表示C原子到Cu(200)表面的垂直距离,Δq表示从Cu(200)表面到C原子的电荷转移

位置 EA(eV) dCu-Cu(Å) dCu-C(Å) h(Å) Δq(e)Hollow 6.22 2.65 1.90 0.29 0.77 Bridge 4.12 2.69 1.80 1.20 0.44
C原子在Cu(200)表面吸附的稳定性,可以通过比较吸附能来确定。吸附能Eads定义为吸附前各部分的总能量减去吸附后体系总能量,其计算表达式如下
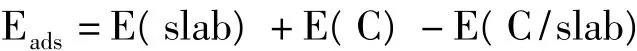
其中E(slab)为未吸附表面的总能量,E(C)为单个C原子的能量,E(C/slab)为C原子吸附后表面的总能量。如表1所列,C原子在Cu(200)表面Hollow位和Bridge位的吸附能分别为6.22 eV和4.12 eV,这说明C原子在Cu(200)表面的吸附为放热过程,C原子可以稳定吸附在Cu(200)表面。C原子在Hollow位的吸附能要大于在Bridge位的吸附能,这说明C原子在Hollow位的吸附更加稳定。
当C原子吸附在Cu(200)表面时,由于C原子和Cu原子具有不同的电负性,在C原子和Cu表面之间将发生电荷转移。由表1可以看到,Δq为正值,这表明吸附的C原子为电子受体,电荷由Cu表面转移到C原子上,当C原子吸附在表面的Hollow位时,由Cu表面转移到C原子上的电荷数最多。为简单起见,下面我们只考虑C原子在Cu(200)表面最稳定吸附位置Hollow位的吸附。对于Cu晶体,每一个Cu原子都为电中性。对于Cu(200)表面,电荷将沿着每一个Cu原子层进行重新分布,如表2所列。C原子吸附后,电荷又将重新分布,电荷由Cu表面转移到C原子上,距离吸附的C原子越近,Cu原子的电荷变化越明显,而远离吸附位置的Cu原子的电荷几乎没有变化。
表2 C原子吸附在Hollow位前后Cu(200)表面的Mulliken电荷分布,表中只给出表面前3个Cu原子层的电荷分布。表中1个数据表明该层的四个Cu原子的带电量相同,4个数据按距离吸附C原子由近及远顺序给出该层四个Cu原子的带电量。
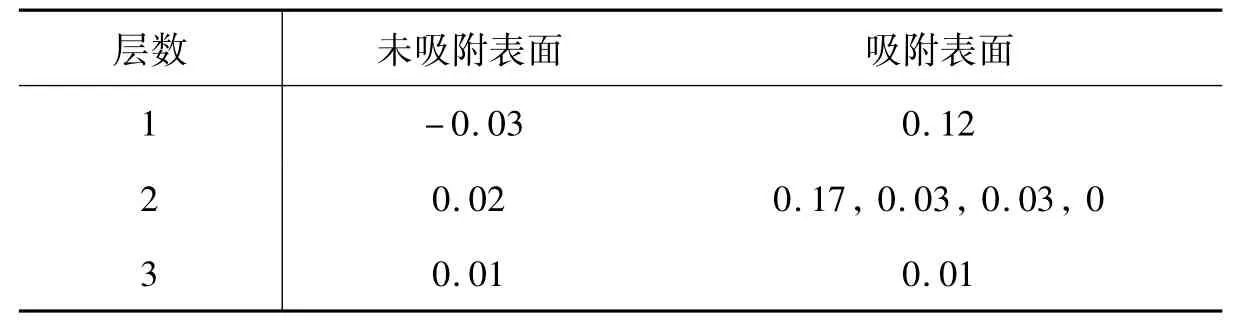
层数 未吸附表面 吸附表面1-0.03 0.12 2 0.02 0.17,0.03,0.03,0 3 0.01 0.01

图2 C原子吸附在Cu(200)表面Hollow位处的电荷差分密度,图中截面为图1(b)中虚线所示的截面,红色为低密度区,蓝色为高密度区,箭头所示为吸附的C原子
图2给出C原子吸附在Cu(200)表面Hollow位处的电荷差分密度图,图中截面通过吸附的C原子和表面的两个Cu原子,如图1(b)中虚线所示。图2中红色为低密度区,蓝色为高密度区。从图中可以看出,在表面层Cu原子处于低电荷密度区,C原子处于高电荷密度区,这说明Cu原子与C原子之间以离子键结合,Cu原子失去电荷,C原子得到电荷,这与前面的Mulliken电荷分布分析是一致的。
3 结论
应用第一原理密度泛函理论计算了C原子吸附后Cu(200)表面的几何结构和电子结构。吸附能的计算结果表明在表面Hollow位C原子的吸附是最稳定的,这是因为在Hollow位时C原子的配位数最大。Mulliken电荷分布和差分电荷密度分析表明C原子吸附后引起Cu(200)表面电荷重新分布,电荷由Cu表面转移到C原子上,C原子与Cu原子之间形成了离子键。本文中Cu表面C原子的吸附研究将为下一步Cu表面石墨烯的生长机理研究提供理论基础。
[1]Novoselov K S,Geim A K,Morozov S V,et al.Electric field effect in atomically thin carbon films[J].Science,2004(306):666-669.
[2]Geim A K,Novoselov K S.The rise of graphene[J].Nat.Mater.,2007(6):183-191.
[3]Soldano C,Mahmood A,Dujardin E.Production,properties and potential of graphene[J].Carbon,2010(48):2127-2150.
[4]Brownson D A C,Kampouris D K,Banks C E.An overview of graphene in energy production and storage applications[J].J.Power Sources,2011(196):4873-4885.
[5]Li X S,Cai W W,An J,et al.Large-area synthesis of high-quality and uniform graphene films on copper foils[J].Science,2009(324):1313-1314.
[6]Li X S,Cai W W,Colombo L,et al.Evolution of graphene growth on Ni and Cu by carbon isotope labeling[J].Nano Lett.,2009(9):4268-4272.
[7]Zhu Y A,Zhou X G,Chen D,et al.First-principles study of C adsorption and diffusion on the surfaces and in the subsurfaces of nonreconstructed and reconstructed Ni(100) [J].J.Phys.Chem.C,2007(111):3447-3453.
[8]Segall M D,Lindan P J D,Probert M J,et al.First-principles simulation:ideas,illustrations and the CASTEP code[J].J.Phys.:Condens.Matter,2002(14):2717-2744.
[9]Perdew J P,Burke K,Ernzerhof M.Generalized gradient approximation made simple[J].Phys.Rev.Lett.,1996(77):3865-3868.
[10]Vanderbilt D.Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J].Phys.Rev.B,1990(41):7892-7895.
