功能化氧化石墨/聚羟基丁酸酯纳米复合材料的制备与性能研究
2014-09-04蔺海兰何飞雄杨海兵龙训利
蔺海兰,卞 军,何飞雄,杨海兵,龙训利
(西华大学材料科学与工程学院特种材料与制备技术重点实验室,四川 成都610039)
生物降解性和生物相容性高分子材料具有环境友好的特征,而且在生物器件中的应用潜力巨大;因此,引起了广大科研人员的研究兴趣。聚羟基丁酸(PHB)是一种来源广泛并可完全降解的热塑性聚酯,作为热塑性可降解聚合物的典型代表,近年来对PHB的改性和应用研究日趋活跃[1],但PHB在熔融加工时热稳定性差,严重限制了其代替传统的不可降解聚合物在工业中的应用[2];因此,对PHB的改性研究主要集中在改善其加工性能以及提高其热稳定性,以拓展PHB的应用范围。目前PHB的改性方法主要有:1)生物法合成一系列含有羟基烷酸单体的共聚物,而不是含有羟基丁酸酯单体;2)将PHB与其他聚合物共混制备相容性聚合物共混物;3)合成含PHB链段的嵌段共聚物。作为一种替代传统PHB改性的有效手段,制备PHB纳米复合材料是一种有效的方法。在纳米复合材料中,纳米填料通过改变基体的结构形态,进而改善聚合物基体本身的性能,甚至赋予材料新的性能[3-4],而且仅需添加少量纳米填料就可以使聚合物的性能获得显著改善。迄今为止,制备PHB基纳米复合材料的填料主要是蒙脱土,大量文献报道了蒙脱土对PHB力学和热性能的影响,然而,PHB/蒙脱土纳米复合材料的主要问题是蒙脱土的添加量过大、基体与蒙脱土的界面结合强度过低,导致复合材料的性能劣化,如密度增大、硬度降低等;因此,制备具有优异力学、热性能和良好的加工性能的PHB纳米复合材料具有重要的研究价值和研究意义。
天然石墨由层状纳米片构成[5]。近年来,聚合物/石墨片纳米复合材料因具有优异的电、热和力学性能而获得广泛的研究[6-7]。因石墨片层间的作用力较弱,一些原子、分子和离子可以插入片层之间,引起石墨片层间距的增大[5],然而,石墨不像层状黏土一样可以通过离子交换反应实现插层,天然石墨片层上无可反应的活性基团;因此,制备完全插层或剥离的聚合物/石墨纳米复合材料较困难。做为石墨的一种典型衍生物,氧化石墨(GO)已经广泛用于替代原始石墨制备聚合物/石墨纳米复合材料[8]。GO是石墨在强氧化剂存在下经深度氧化而得,在氧化过程中可在石墨结构中引入功能性官能团[9]。这些官能团赋予GO片强烈的亲水性,并能在水或碱性溶液中均匀分散且稳定存在[10], 然而GO的强亲水性导致其与一些非极性的聚合物基体相容性变差。为提高GO在非质子极性溶剂(如DMF)中的分散性,以及改善其与聚合物基体的相容性,Stankovich et al[11]采用异氰酸酯对GO的表面进行化学功能化处理,以改善GO片层的表面性质。结果表明,GO的功能化改性促进了其在DMF溶剂中的剥离和分散,并有效改善了其与聚合物基体的复合效果。本课题组也报道了采用“接枝”法将甲苯-2,4-二异氰酸酯(TDI)引入到GO片层的表面以对片层表面进行功能化处理,并将功能化的GO与聚甲基乙撑碳酸酯(PPC)进行共混,获得了高性能的PPC/功能化石墨片纳米复合材料[12]。Xu et al[13]通过对GO进行类似的“接枝”改性,以促进GO在水和有机介质中的分散性,该“接枝”方法是将含有某种末端官能团的聚合物接枝到基体的表面以改变基体的表面性质[14]。
本文报道一种具有优异力学和热性能的PHB/功能化石墨片纳米复合材料。为了避免在传统熔融共混制备过程中PHB发生降解,本研究采用溶液插层法制备高度剥离的PHB/功能化石墨片纳米复合材料,并采用原子力显微镜(AFM)、X线衍射(XRD)和扫描电子显微镜(SEM)对MGO和MGO/PHB纳米复合材料的微观形貌结构进行分析,采用微分量热仪(DSC)和拉伸测试对MGO/PHB纳米复合材料的热性能和力学性能进行测试表征。
1 实验部分
1.1 原材料
聚羟基丁酸酯(PHB):Sigma-Aldrich 化学品公司。天然石墨粉(NGP,SP-2,C含量 > 99%,D=5μm):青岛天和石墨有限公司。高锰酸钾(KMnO4)、浓硫酸(>96%)、双氧水、 1,4-丁二醇(BD)、丙酮、 乙醇、N,N-二甲基甲酰胺 (DMF) 和甲苯-2,4-二异氰酸酯(TDI):成都科隆试剂有限公司。
1.2 GO,GO-TDI 和 MGO (GO-TDI-BD)的制备
本研究使用的GO是在KMnO4和 H2SO4存在下经深度氧化而得,具体过程见文献所述[15]。GO-TDI和MGO (GO-TDI-BD)的制备如图1所示。典型的制备过程如下:先将2 g GO加入含有100 mL DMF的250 mL 三颈瓶中,氮气保护下经磁力搅拌获得均匀的GO/DMF分散体系;再将2 g TDI加入上述体系中于80 ℃ 下磁力搅拌反应 24 h后,将过量BD (5 g) 加入上述体系中并于80 ℃ 下继续反应 24 h,将该分散液倒入过量的丙酮中进行沉淀,沉淀物经离心浓缩和水洗以除去未反应的BD;最后,所得产物(MGO) 在100 ℃ 下真空干燥48 h备用。
1.3 MGO/PHB 纳米复合材料的制备
采用溶液插层法制备MGO/PHB纳米复合材料。为了获得稳定且均匀分散的剥离石墨纳米片,首先将定量的MGO加入25 mL DMF中并超声波分散获得MGO/DMF分散液。同时,将定量的PHB溶解于300 mL DMF中,然后将其与MGO/DMF分散液混合,并经超声波分散4 h直至获得稳定、均匀的体系,以完成插层复合过程。再将其缓慢加入到大量的乙醇溶液中,并激烈搅拌。将沉淀物浓缩、过滤并于80 ℃下真空干燥24 h后得到MGO/PHB纳米复合材料粉末,所得粉末在200 ℃下经热压成薄片进行分析测试。 MGO/PHB纳米复合材料的合成技术路线如图1所示。
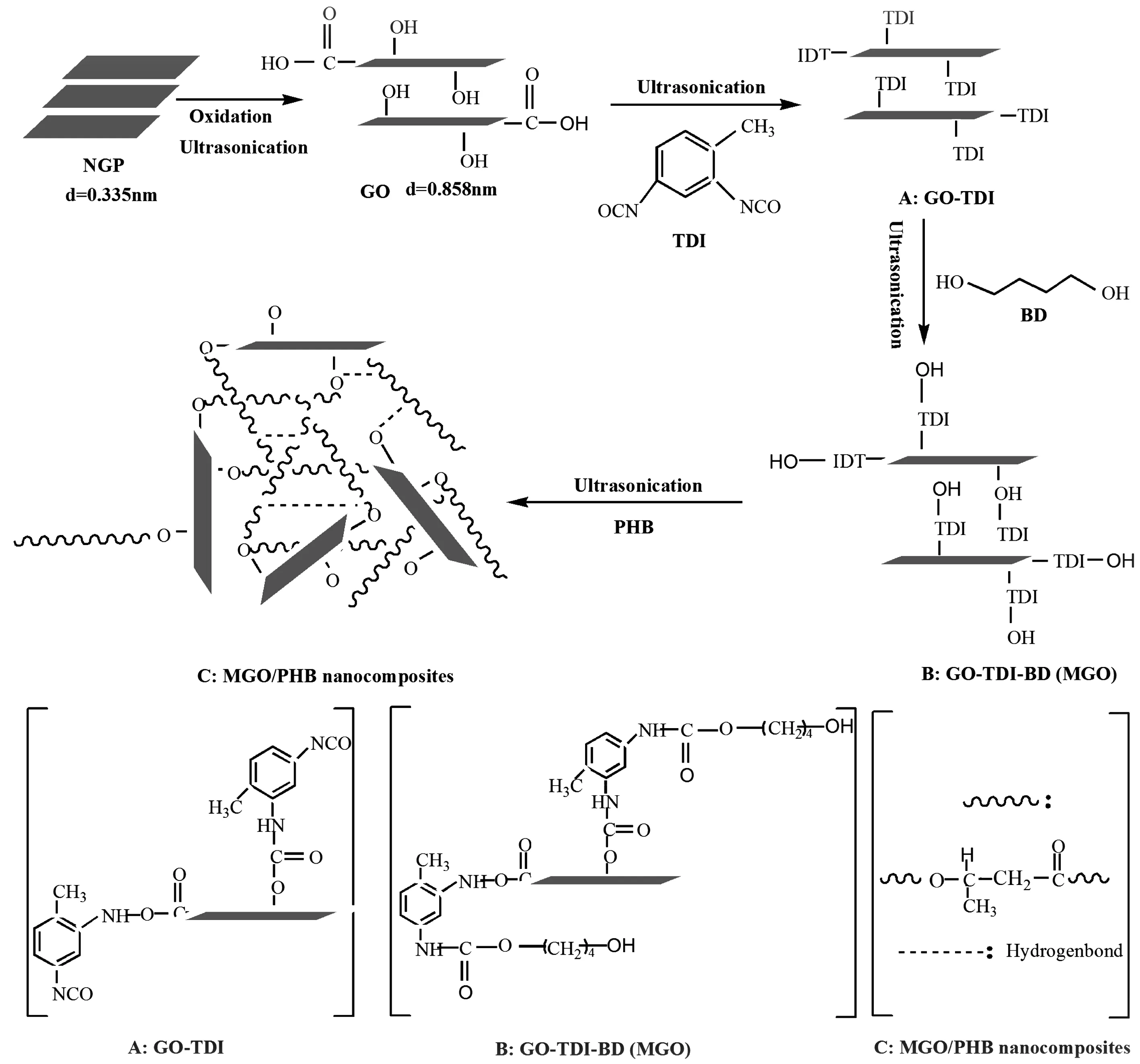
图1 MGO/PHB 纳米复合材料的合成技术路线
1.4 分析测试与表征
1.4.1 形态与结构表征
采用JSM-6510LV型(日本电子美国公司)扫描电子显微镜(SEM)对样品的微观形貌与结构进行表征,SEM测试前样品表面先经喷金处理。采用带有敲击模式的原子力显微镜(AFM)技术表征MGO的表面形貌,AFM测试前先将MGO均匀分散于DMF中,然后将MGO/DMF分散体液滴滴于金属样品台上,经充分干燥后测试。
1.4.2 X线衍射分析(XRD)
采用Rigaku D/max-1200X 型X线分析仪(40 kV, 200 mA, Cu Ka, λ = 0.154 nm)对样品的结构进行测试。扫描在室温下进行,扫描角度为1.5°到 60° ,步长为1.2(°) /min,NGP、GO和 MGO 样品为粉末形式,MGO/PHB 纳米复合材料为热压所得薄片。
1.4.3 微分扫描量热仪分析(DSC)
采用NETZSCH DSC-200PC型微分扫描量热仪于N2气氛下对MGO/PHB 纳米复合材料的熔融和结晶行为进行分析测试。先将样品(5~10 mg)从-50oC加热到250 ℃,恒定5 min,再降温到-50 ℃,恒温5 min,最后再次升温到250 ℃,升温和降温速率都为10 ℃/min。在升、降温过程中记录DSC曲线,并通过DSC曲线获得纳米复合材料的热行为参数。复合材料的相对结晶度按下式计算:
试中:Xc为相对结晶度;ΔHc为结晶潜热;ΔHf为结晶度为100%的结晶PHB的熔融热焓(ΔHf= 146 J/g );Wf为MGO在复合材料中的质量分数。
1.4.4 拉伸性能测试
采用INSTRON3365 型电子拉力机测试复合材料的拉伸性能。测试前将MGO/PHB纳米复合材料热压膜裁成62.5 × 3.25 × 0.7 mm3(长 × 宽 ×厚)的哑铃型试样,拉伸测试速度为10 mm/min,测试温度为室温,每个配比测试5个试样并取平均值。
2 结果与分析
2.1 MGO、MGO/PHB纳米复合材料的结构与形貌

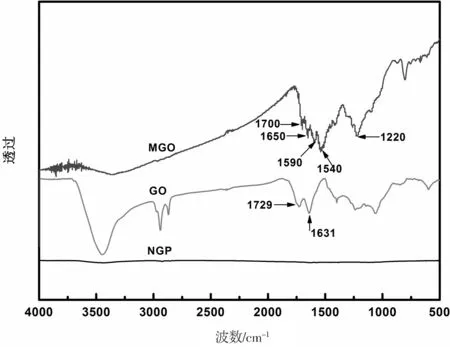
图2 NGP、GO和MGO的红外光谱图
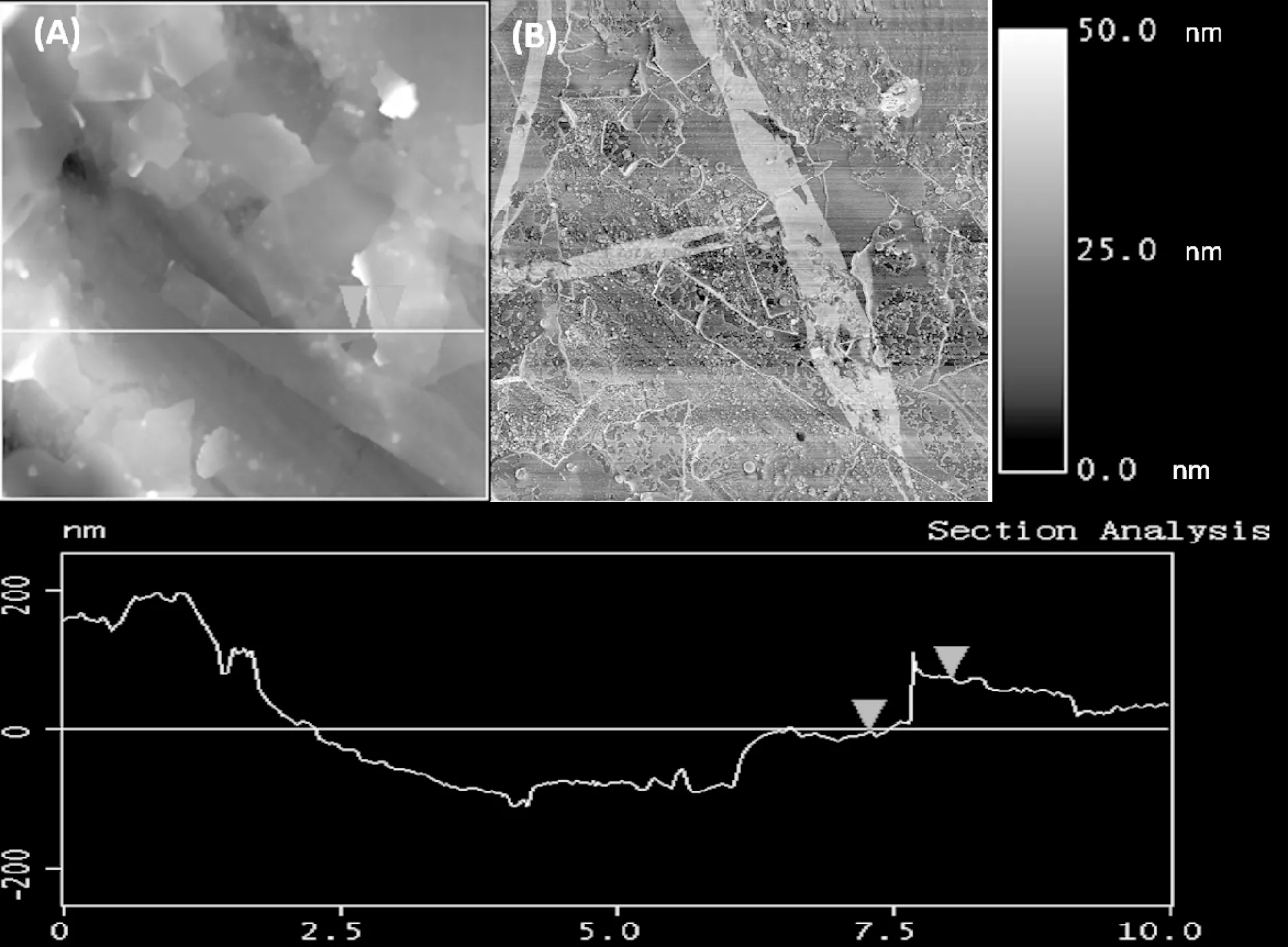
(A):高度曲线;(B):相图
图4(A)和(B)分别表示NGP、GO、GO-TDI、GO-TDI-BD和不同MGO质量分数的MGO/PHB纳米复合材料的X线衍射曲线。在衍射角2θ为26.6°(002,d=0.335 nm)处出现的衍射峰是NGP的特征衍射峰(曲线(a))。经强氧化剂氧化后,在衍射角2θ为12.1°处出现GO的特征衍射峰,归结为GO的(001)衍射峰,层间距d=0.858 nm(曲线(b))。对于MGO,在2θ为8.8°处出现衍射峰,相应的层间距增大到d=1.3 nm(曲线(d))。层间距增大主要是由于TDI和BD已经成功接枝到GO的表面引起的。有趣的是,研究发现GO经功能化改性后,MGO在水中不能均匀分散,长时间静置后有沉淀产生。这一现象表明石墨片的表面性质已由亲水性转变为疏水性,这种转变无疑对与非极性聚合物的插层共混是有利的。从图4(B)可以看出,当MGO和PHB复合后,MGO的001衍射峰强度逐渐减弱并最终消失 (见图4(B)中插入的曲线图),表明MGO的有序性结构被破坏。这主要是因为在材料复合过程中,PHB分子链与疏水性MGO的表面产生相互作用,PHB分子链插入MGO片层之间导致MGO的层间距变大,并最终引起MGO的剥离[16]。
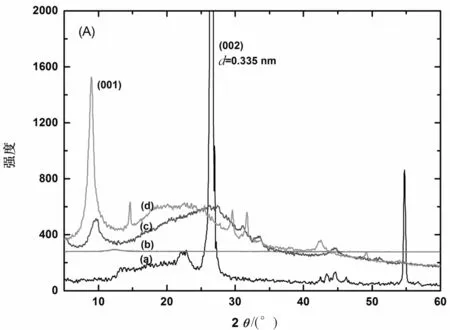
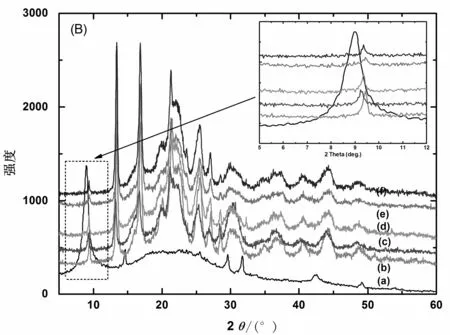
(A): NGP (a), GO (b), GO-TDI (c), MGO (d); (B): MGO (a) 和不同MGO质量分数的MGO/PHB 纳米复合材料 (b-g) (插入图片表示2θ为5°~12°范围内的衍射模式)
图4 XRD衍射曲线
微观结构对复合材料的性能有重要影响。在本研究中,先用TDI对GO进行功能化处理以在GO表面引入活性位,获得GO-TDI,再通过“接枝”法将GO-TDI用BD功能化处理,以便在其表面引入羟基。这种表面功能化处理的GO表面可以作为共混过程中PHB的“接枝点”,这些“接枝点”能有效改善PHB与填料石墨之间的界面黏结强度,并提高2者的相容性。为表征MGO在PHB中的分散性,采用SEM对纯PHB和MGO/PHB纳米复合材料的表面进行观察,如图5所示。从图5(A)可以看出,纯PHB表现出光滑的断面特征,表明PHB是脆性断裂。而对于MGO/PHB 纳米复合材料,随着MGO的逐渐添加,复合材料的断面逐渐变得粗糙,如图5(B)—(F)所示。进一步研究发现,尽管复合材料断面随着MGO添加量的增加而越来越粗糙,但即使MGO的质量分数为4.0%时,也并未观察到明显的MGO团聚区域,表明MGO已经在PHB基体中获得了良好的剥离和分散,这归功于复合过程中基体PHB与MGO之间的良好界面相互作用。同时,SEM也证实了本研究所采用的“接枝”方法的有效性。填料在聚合物基体中的分散性能直接决定其对基体力学性能、热性能等的改善效果。除此之外,复合材料的性能也受填料的长宽比、表面体积比等参数的影响。本研究获得的MGO具有较高的长宽比和在基体中的良好分散性,这些因素将不同程度地提高复合材料的性能,下面的研究将进一步对此进行证实。

(A):纯 PHB; (B):0.25%;(C):0.5%;(D):1.0%;(E):2.0%;(F):4.0%。
2.2 PHB和MGO/PHB纳米复合材料的热性能
采用DSC研究了PHB和MGO/PHB纳米复合材料的热性能,并记录了DSC曲线,如图6所示。从曲线可以获得熔融温度(tm)、起始和终止结晶温度 (tc,o和tc,e)、结晶峰温度(tc,p)和熔融和结晶热焓(ΔHm和ΔHc)等参数,如表1所示。从DSC加热曲线(图6(A))可以看出,PHB中加入MGO后,PHB的熔融峰值向高温方向移动。从DSC加热曲线(图6(B))可以看出,加入MGO后,PHB的结晶峰也逐渐向高温方向移动,这归于MGO在PHB基体中的异相成核效应,而且这种成核效应在高MGO质量分数时更为明显。进一步研究发现,复合材料的相对结晶度(Xc)和过冷度(定义为熔融峰温度值与结晶峰温度值之差,Δt=tm,p-tc,p)都比纯PHB高。复合材料的结晶温度和熔融温度的改善表明将PHB与MGO进行纳米复合是提高PHB热稳定性的有效方式。
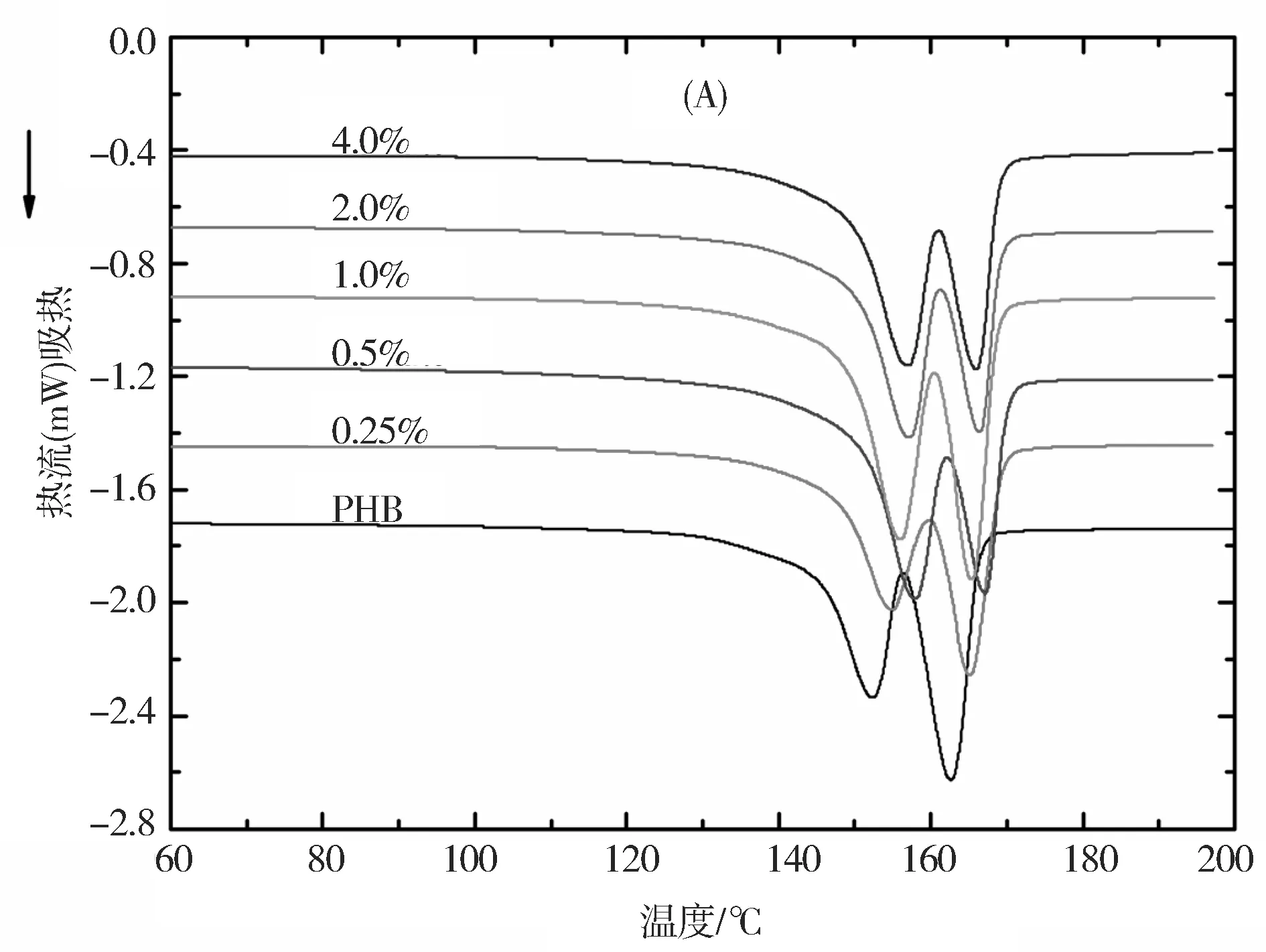
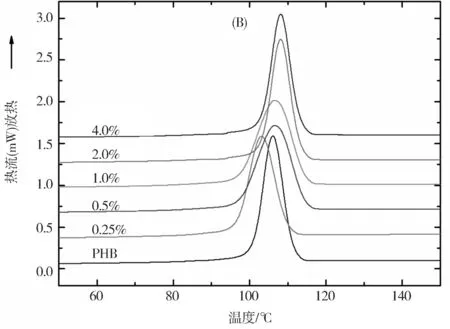
图6 纯PHB和不同MGO质量分数的MGO/PHB纳米复合材料的DSC升温曲线(A)和降温曲线(B)
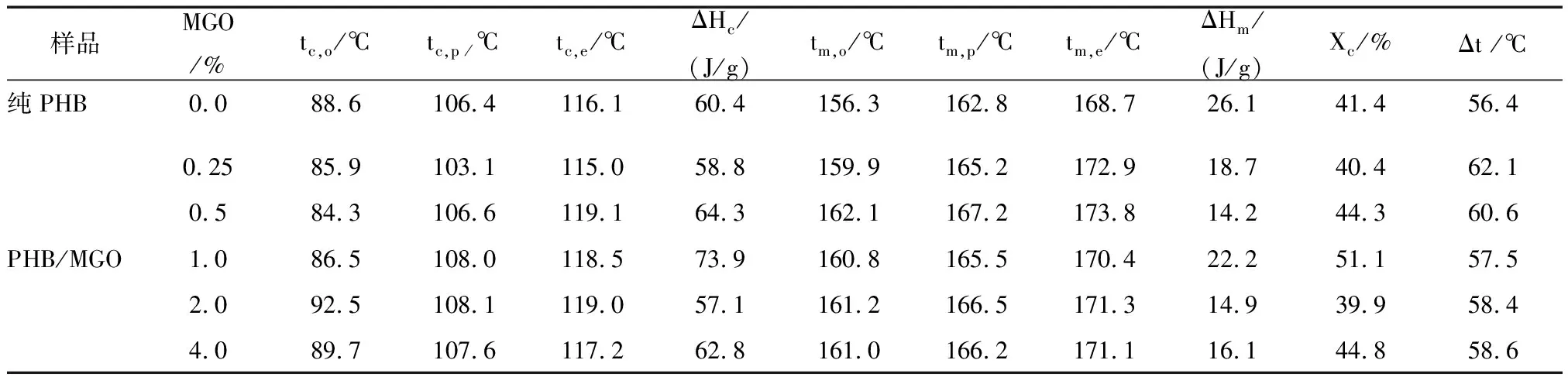
样品MGO/%tc,o /℃tc,p /℃tc,e /℃ΔHc/(J/g)tm,o /℃tm,p /℃tm,e /℃ΔHm/(J/g)Xc /%Δt /℃纯PHB0.088.6106.4116.160.4156.3162.8168.726.141.456.40.2585.9103.1115.058.8159.9165.2172.918.740.462.10.584.3106.6119.164.3162.1167.2173.814.244.360.6PHB/MGO1.086.5108.0118.573.9160.8165.5170.422.251.157.52.092.5108.1119.057.1161.2166.5171.314.939.958.44.089.7107.6117.262.8161.0166.2171.116.144.858.6
注:tc,o为起始结晶温度;tc,p为结晶峰温度;tc,e为终止结晶温度;ΔHc为结晶热焓;tm,o为起始熔融温度;tm,p为熔融峰温度;tm,e为终止熔融温度;ΔHm为熔融热焓;Xc为相对结晶度;Δt为过冷度 (tm,p-tc,p)。
2.3 MGO/PHB纳米复合材料的拉伸性能
本文测试了MGO/PHB纳米复合材料的拉伸性能,并以断裂强度和断裂模量作为力学性能的关键指标讨论MGO质量分数对复合材料力学性能的影响。拉伸过程的应力-应变曲线如图7(A)所示,图7(B)为复合材料及其拉伸试样的数字照片,图7(C)为MGO质量分数对复合材料断裂强度和断裂模量的影响曲线。
图7(A)表明,与纯PHB相比,随MGO添加量的逐渐增加,复合材料的断裂伸长率逐渐降低,说明复合材料的脆性变大。尽管如此,复合材料仍然能热压成膜并能裁剪成规则哑铃型试样,如图7(B)所示。从图7(C)可以看出,MGO/PHB纳米复合材料的断裂强度在MGO的质量分数低于2.0%时有逐渐降低的趋势,而当MGO质量分数大于2.0%时断裂强度逐渐变大;然而,随着MGO质量分数的逐渐增加,MGO/PHB纳米复合材料的断裂模量表现出与断裂强度不同的变化规律,特别是当MGO质量分数低于0.5%时最为明显,但当MGO质量分数处于0.5%~1.0%时断裂强度则逐渐增加,而超过1.0%后增大明显,因此,MGO/PHB纳米复合材料的断裂模量与断裂强度出现增大的临界MGO质量分数在1%~2.0%之间。据文献报道[17],力学性能随填料含量的变化出现的临界点与填料在基体中完全形成三维网络时填料的添加临界值(MGO添加量的阀值)具有某种关联,即在该临界值时填料三维网络完全形成。进一步研究发现,在所研究的MGO用量范围内,MGO/PHB 纳米复合材料都表现出较纯PHB更为优异的断裂模量值。当MGO的质量分数为4.0%时,MGO/PHB 纳米复合材料的断裂模量为139.5 MPa,比纯PHB(78.2 MPa)提高了61.3 MPa。可见,添加MGO能明显改善复合材料的力学性能,这是由于采用“接枝”法在GO表面引入大量活性官能团。这些官能团与PHB分子链之间的强相互作用不仅能改善PHB与MGO的界面结合强度,还能促进MGO的剥离和分散。当MGO的质量分数较低时,借助于超声波的分散作用,其能均匀分散于基体中,尽管复合材料的断裂伸长率与纯PHB相比有所降低,但拉伸过程中仍能产生较大的变形(如图7(A)所示)。同时,复合材料的断裂强度仍保持较高值(如图7(C)所示)。随MGO质量分数逐渐增加,复合材料的断裂强度呈现逐渐下降到一最低值,并保持不变,之后逐渐增大的趋势,而复合材料的断裂模量随MGO质量分数逐渐变大。一方面,MGO 在PHB中均匀分散导致石墨烯纳米片层的比表面积增大,对PHB的分子链具有吸附作用,从而限制了PHB的分子链运动;另一方面,MGO与PHB的分子链之间存在强的化学作用力,这2个方面的原因都导致复合材料的力学性能得以改善。
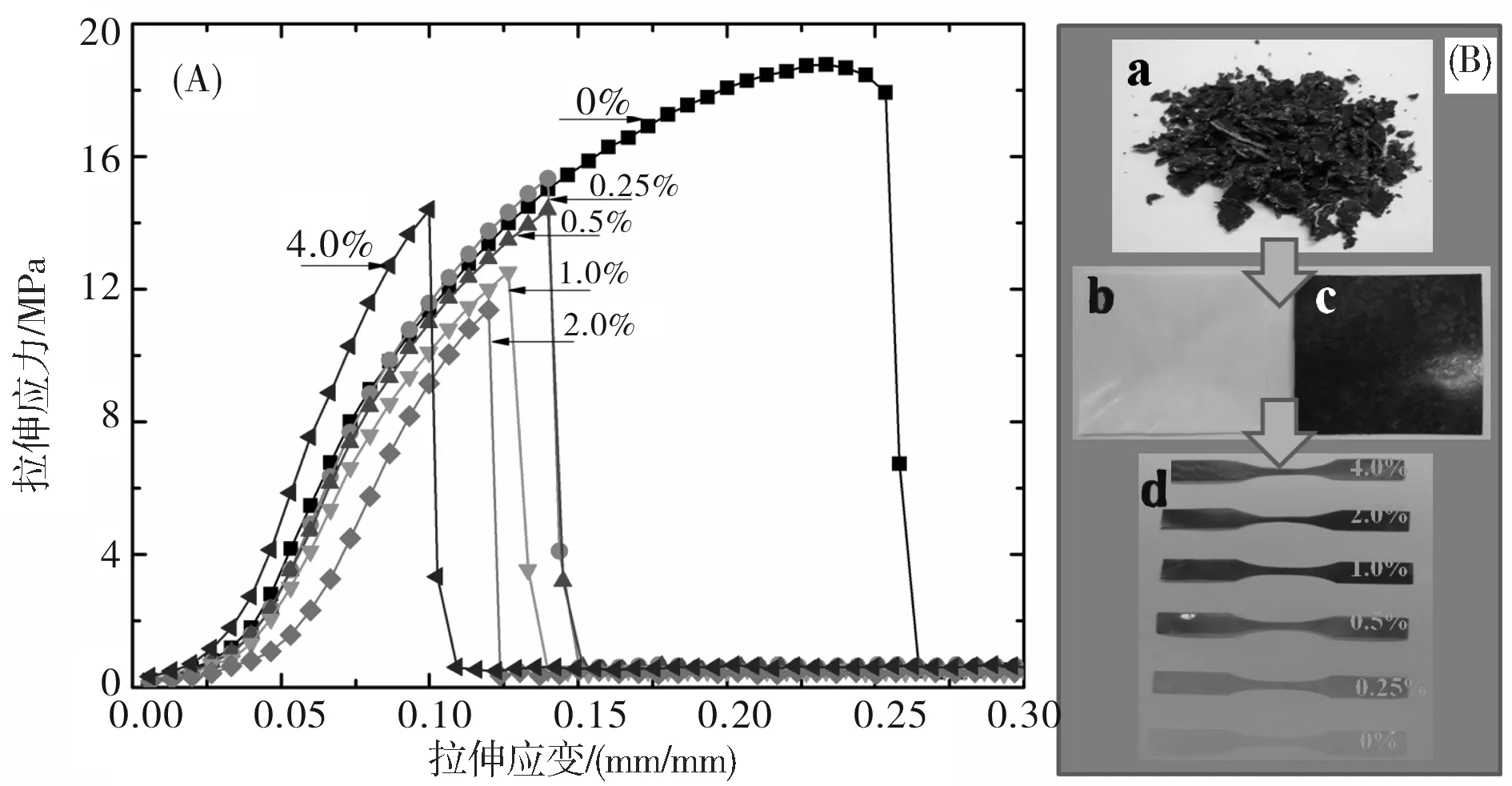
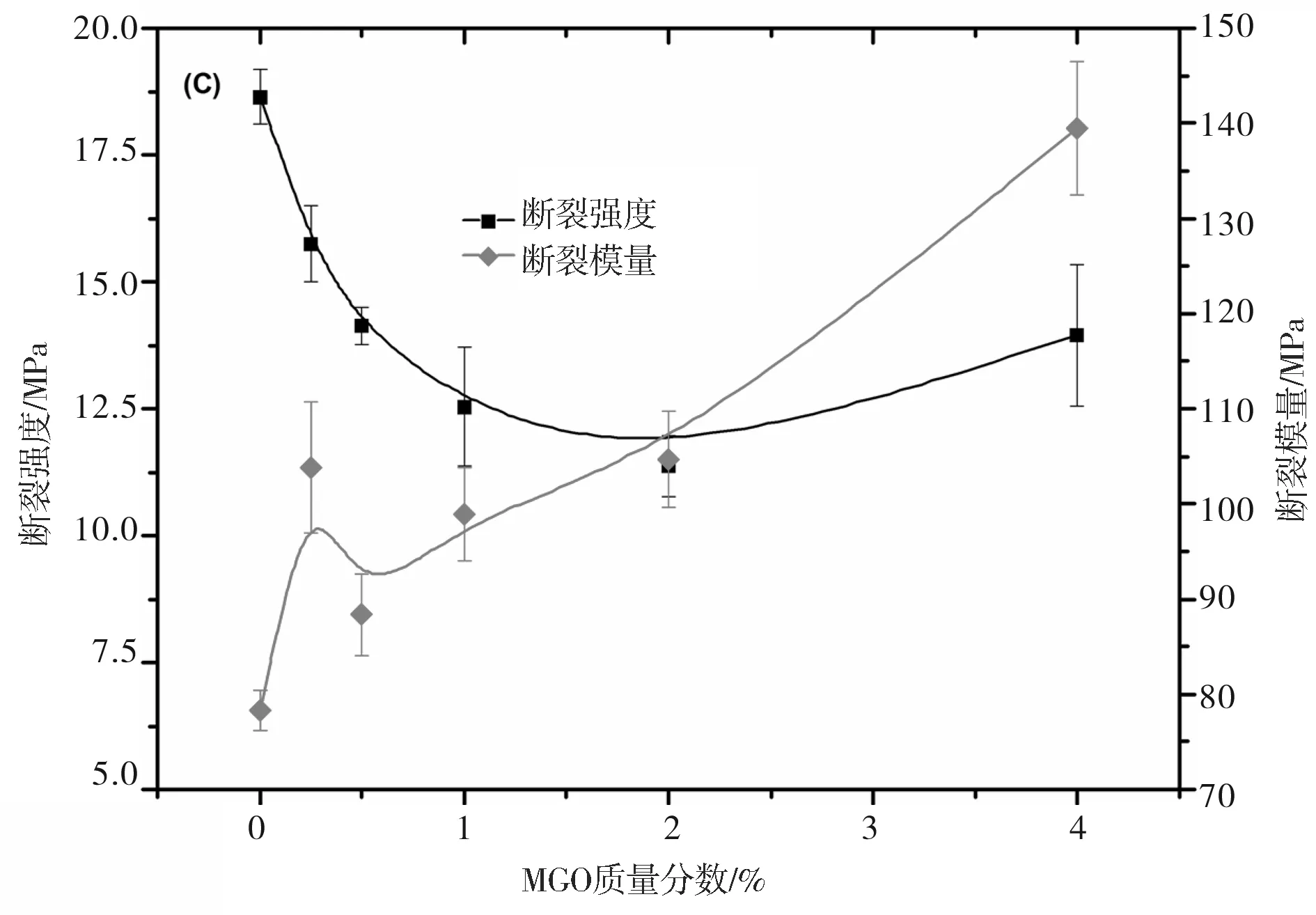
图7 纯PHB和不同MGO含量的MGO/PHB纳米复合材料的力学性能测试结果
图7中:(A)为应力-应变曲线;(B)为样品的数字照片(a为MGO质量分数为1.0%时MGO/PHB复合材料经过乙醇沉淀后得到的粉末样品,b为纯PHB热压膜,c为MGO质量分数为1.0%时MGO/PHB复合材料的热压膜,d为不同MGO质量分数的MGO/PHB复合材料的拉伸哑铃型试样);(C)为不同MGO质量分数对MGO/PHB复合材料的断裂强度和断裂模量的影响。
3 结论
本文采用溶液插层技术成功制备了一种新型MGO/PHB纳米复合材料。GO的化学功能化促进了其与PHB基体的液相复合效果。PHB分子链已经插入到MGO片层之间,导致石墨片层间距增大,并促进了MGO在基体PHB中的剥离和均匀分散。与纯PHB相比,因MGO在PHB基体中的异相成核作用,导致复合材料的结晶速率变快、结晶度增大。随着MGO质量分数逐渐增加,复合材料的断裂强度先降低后增大,而含不同MGO质量分数的复合材料都表现出更高的断裂模量值。复合材料的力学性能变化与MGO添加量的阀值有一定关系。总之,本文提供了一种制备可降解PHB基纳米复合材料的有效技术方法,PHB基体性能在添加MGO后获得明显改善,这拓宽了PHB的应用领域。
[1]Fernandes E G, Pietrini M, Chiellini E. Thermo-mechanical and Morphological Characterization of Plasticized Poly [(R)-3-hydroxybutyric Acid] [J]. Macrom Symp, 2004, 218: 157-164.
[2] Lee S N, Lee M Y, Park W H. Thermal Stabilization of Poly(3-hydroxybutyrate) by Poly(glycidyl Methacrylate) [J]. J Appl Polym Sci, 2002, 83: 2945-2952.
[3] Woodruff M A, Hutmacher D W. The Return of a Forgotten Polymer-polycaprolactone in the 21st Century [J]. Prog Polym Sci, 2010, 35 (10): 1217-1256.
[4] Hui Lian C, Muthunarayanan M, Youngkeun A, et al. Biodegradable Particulate Delivery of Vascular Endothelial Growth Factor Plasmid from Polycaprolactone/Polyethylenimine Electrospun Nanofibers for the Treatment of Myocardial Infarction [J]. J Nanosci Nanotechnol, 2011, 11 (8): 7307-7313.
[5] Chung D D L. Review Graphite [J]. J Mater Sci, 2002, 37 (8): 1475-1489.
[6] Zhang M, Li D, Wu D, et al. Poly(ethylene terephthalate)/expanded Graphite Conductive Composites: Structure, Properties, and Transport Behavior [J]. J Appl Polym Sci ,2008, 108 (3): 1482-1489.
[7] Kai W H, Hirota Y, Hua L, et al. Thermal and Mechanical Properties of a Poly(-caprolactone)/Graphite Oxide Composite [J]. J Appl Polym Sci ,2008, 107 (3): 1395-400.
[8] Matsuo Y, Miyabe T, Fukutuka T, et al. Preparation and Characterization of Alkylamine-intercalated Graphite Oxides [J]. Carbon ,2007,45 (5): 1005-1012.
[9] He H, Klinowski J, Forster M, et al. A New Structural Model for Graphite Oxide [J]. Chem Phys Lett, 1998, 287 (1/2): 53-56.
[10] Hirata M, Gotou T, Ohba M. Thin-film Particles of Graphite Oxide. 2: Preliminary Studies for Internal Micro Fabrication of Ssingle Particle and Carbonaceous Electronic Circuits [J]. Carbon, 2005,43 (3): 503-510.
[11] Stankovich S, Dikin D A, Dommett G H B, et al. Graphene-based Composite Materials [J]. Nature ,2006, 442 (7100): 282-286.
[12] Bian J, Wei X W, Lin H L, et al. Preparation and Characterization of Modified Graphite Oxide/Poly(propylene carbonate) Composites by Solution Intercalation [J]. Polym Degrad Stab, 2011, 96 (10): 1833-1840.
[13] Xu C, Wu X, Zhu J, et al. Synthesis of Amphiphilic Graphite Oxide [J]. Carbon, 2008, 46 (2): 386-389.
[14] Ranjan R, Brittain W. Combination of Living Radical Polymerization and Click Chemistry for Surface Modification[J]. Macromolecules, 2007,40 (17): 6217-6223.
[15] Bian J, Xiao M, Wang S J, et al. Highly Effective Synthesis of Dimethyl Carbonate from Methanol and Carbon Dioxide Using a Novel Copper-nickel/graphite Bimetallic Nanocomposite Catalyst [J]. Chem Eng J ,2009, 147 (2-3): 287-296;
[16] Shi X D, Gan Z H. Preparation and Characterization of Poly(propylene carbonate)/montmorillonite Nanocomposites by Solution Intercalation [J]. Eur Polym J ,2007,43 (12): 4852-4856.
[17] Thongruang W, Spontak R J, Balik C M. Correlated Electrical Conductivity and Mechanical Property Analysis of High-density Polyethylene Filled with Graphite and Carbon Fiber [J]. Polymer ,2002, 43 (8): 2279-2286.
