四氢喹啉-4-醇类衍生物的合成*
2014-08-30陈永正郑代军刘小卒周晓建
陈永正,郑代军,刘小卒,周晓建,徐 可
(遵义医学院 药学院,贵州 遵义 563000)
·研究简报·
四氢喹啉-4-醇类衍生物的合成*
陈永正,郑代军,刘小卒,周晓建,徐 可
(遵义医学院 药学院,贵州 遵义 563000)
以1,2,3,4-四氢喹啉为初始原料,经N-酰基保护、氧化、脱保护、还原等反应合成了一系列1,2,3,4-四氢喹啉-4-醇类的衍生物,总产率17%~41%,其结构经1H NMR和13C NMR确证。
四氢喹啉;氨基醇;四氢喹啉-4-醇;合成
1,2,3,4-四氢喹啉类化合物是一类重要的杂环化合物,类似结构广泛存在于部分天然生物碱中[1],在药物[2]、染料[3]及作为催化剂配体[4]进行不对称合成等方面有着广泛应用。四氢喹啉类化合物的合成方法主要有非饱和体系的还原[5]、直链体系的关环[6]、苯胺和两分子醛的缩合[7]、N上芳香化的亚甲基亚胺正离子和烯烃的缩合[8]、环的重排等。1949年,William P等[9]以苯胺和丙烯酸甲酯为原料经1,4-加成和傅克酰基化反应制得4-羰基-1,2,3,4-四氢喹啉。
本文以1,2,3,4-四氢喹啉(1)为起始原料,经N-酰基保护、氧化[10]、脱保护、还原等反应合成了一系列1,2,3,4-四氢喹啉-4-醇类衍生物[11](4a~4c和7~9,Scheme 1),总收率17%~41%,其结构经1H NMR和13C NMR确证。
1 实验部分
1.1 仪器与试剂
ZF-2型三用紫外分析仪;Bruker 300MHz型核磁共振仪(DMSO-d6为溶剂,TMS为内标)。
所用试剂均为分析纯。
1.2 合成
(1)N-叔丁氧羰基四氢喹啉(2a)的合成
在圆底烧瓶中依次加入12.66g(20mmol),二氯甲烷20mL及三乙胺3.03g(30mmol),搅拌下于室温滴加二碳酸二叔丁酯[(Boc)2O]5.56g(25.5mmol),滴毕,回流(40℃)反应10h。用水洗涤、无水Na2SO4干燥,减压浓缩后经硅胶柱层析[洗脱剂:A=V(乙酸乙酯)∶V(石油醚)=1∶10]纯化得黄色油状液体2a4.56g,产率98%;1H NMR(CDCl3)δ: 6.95~7.65(m,4H),3.71(t,J=6.0Hz,2H),2.76(t,J=6.6Hz,2H),1.88~1.96(m,2H),1.52(s,9H);13C NMR(CDCl3)δ: 153.9,138.6,129.9,128.5,125.6,124.1,123.2,80.6,44.6,27.4,27.4,23.5。
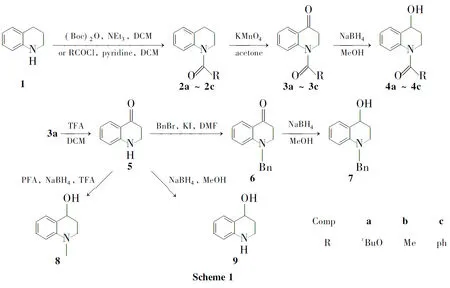
(2)N-乙酰基四氢喹啉(2b)的合成
在反应瓶中依次加入15mL的二氯甲烷(150mL)溶液和吡啶5mL,冰浴冷却,搅拌下缓慢滴加乙酰氯3.54mL,滴毕,于室温反应30min。加水(5mL)淬灭反应,分液,有机相用无水硫酸钠干燥,减压浓缩后经硅胶柱层析(洗脱剂:A=1∶10)纯化得黄色液体2b6.88g,产率98%;1H NMR(CDCl3)δ: 7.09~7.26(m,4H),3.78(t,J=6.5Hz,2H),2.71(t,J=6.5Hz,2H),2.22(s,3H),1.90~1.99(m,2H);13C NMR(CDCl3)δ: 170.1,128.4,126.0,125.1,124.5,26.8,24.0,23.1。
(3)N-苯甲酰基四氢喹啉(2c)的合成
在反应瓶中依次加入10.67mL的二氯甲烷(25mL)溶液和吡啶0.48mL,搅拌下于室温缓慢滴加苯甲酰氯0.70mL,滴毕,反应15min。加水淬灭反应,分液,有机相用无水硫酸钠干燥,减压浓缩后经硅胶柱层析(洗脱剂:A=1∶10)纯化得黄色固体2c0.64g,产率50%;1H NMR(CDCl3)δ: 7.24~7.37(m,5H),7.15(d,J=7.5Hz,1H),5.49(t,J=7.4Hz,1H),6.86(t,J=7.8Hz,1H),6.72(d,J=7.8Hz,1H),3.91(t,J=6.5Hz,2H),2.84(t,J=6.6Hz,2H),2.01~2.10(m,2H);13C NMR(CDCl3)δ: 170.3,139.3,136.3,131.6,130.1,128.6,128.3,128.0,125.7,125.4,124.5,44.4,26.9,24.1。
(4)3的合成(以3a为例)
在圆底烧瓶中依次加入2a4.56g,丙酮120mL,15%MgSO4溶液20mL和KMnO49.48g(60mmol),搅拌下于室温反应36h。加水100mL,用Na2SO3淬灭反应,过滤,滤液用乙酸乙酯萃取,有机相用无水硫酸钠干燥,减压浓缩后经硅胶柱层析(洗脱剂:A=1∶10)纯化得白色固体3a2.73g。
用类似方法合成3b和3c。
3a:产率55%;1H NMR(CDCl3)δ: 7.13~8.00(m,4H),4.16(t,J=6.0Hz,2H),2.77(t,J=6.0Hz,2H),1.55(s,9H);13C NMR(CDCl3)δ: 194.2,152.7,144.1,133.9,127.3,124.9,123.9,123.7,82.2,44.3,39.0,28.3。
3b(反应12h):产率49%;1H NMR(CDCl3)δ: 8.00(d,J=7.4Hz,1H),7.43~7.57(m,2H),7.26(t,J=8.0Hz,1H),4.23(t,J=6.2Hz,2H),2.79(t,J=6.2Hz,2H),2.33(s,3H);13C NMR(CDCl3)δ: 193.9,169.3,143.9,134.0,127.7,126.0,125.5,124.1,43.9,39.5,23.1。
3c(反应7h):产率50%;1H NMR(CDCl3)δ: 8.01(d,J=7.8Hz,1H),7.13~7.50(m,7H),6.91(d,J=8.2Hz,1H),4.33(t,J=12.6Hz,2H),2.88(t,J=6.4Hz,2H);13C NMR(CDCl3)δ: 193.7,170.1,144.5,135.0,133.6,131.1,128.6,128.5,127.6,124.9,124.8,124.5,45.2,39.6。
(5)4的合成(以4a为例)
在反应瓶中加入3a250mg(1mmol)的甲醇(10mL)溶液和NaBH460mg,搅拌下于室温反应30min。加水淬灭反应,用乙酸乙酯萃取,合并萃取液,用无水硫酸钠干燥,减压浓缩后经硅胶柱层析(洗脱剂:A=1∶2)纯化得白色固体4a0.16g。
用类似方法合成4b和4c。
4a:产率65%;1H NMRδ: 6.93~7.61(m,4H),5.38(d,J=3.9Hz,1H),4.54(d,J=3.9Hz,1H),3.74~3.80(m,1H),3.50~3.57(m,1H),1.91~1.99(m,1H),1.73~1.78(m,1H),1.44(s,9H);13C NMRδ: 152.9,137.1,132.6,127.9,126.7,123.0,122.8,80.3,64.1,40.9,32.2,27.9。
4b:产率86%,1H NMRδ: 7.11~7.50(m,4H),5.44(d,J=5.4Hz,1H),4.55~4.58(m,1H),4.02~3.85(m,1H),3.48~3.54(m,1H),2.16(s,3H),2.02~2.13(m,1H),1.71~1.77(m,1H);13C NMRδ: 169.2,137.3,127.0,126.7,124.1,123.9,64.2,32.9,23.4。
4c: 产率71%;1H NMRδ:7.44~7.34(m,6H),7.08~7.03(m,1H),6.97~6.95(m,1H),6.80(d,J=8.1Hz,1H),5.52(d,J=5.4Hz,1H),4.72~4.70(m,1H),3.92~3.88(m,1H),3.68~3.63(m,1H),2.17~2.15(m,1H),1.87~1.83(m,1H);13C NMRδ: 169.4,137.5,136.5,134.2,130.1,128.2,128.1,127.4,126.4,124.4,124.1,64.1,41.8,32.8。
(6)2,3-二氢喹啉-4(1H)-酮(5)的合成
在反应瓶中依次加入3a0.5g(2mmol),二氯甲烷20mL和三氟乙酸(TFA)0.9mL(12mmol),搅拌下回流(40℃)反应3h。用1mol·L-1NaOH溶液调至pH9,用二氯甲烷(3×10mL)萃取,合并萃取液,用无水硫酸钠干燥,减压浓缩后经柱层析(洗脱剂:A=1∶4)纯化得绿色黏稠液体50.25g,产率85%;1H NMR(CDCl3)δ: 6.65~7.85(m,4H),4.49(s,1H),3.56(t,J=6.6Hz,2H),2.69(t,J=6.9Hz,2H);13C NMR(CDCl3)δ: 193.7,152.0,135.1,127.6,119.3,117.8,115.8,42.2,38.0。
(7)1-苄基-1,2,3,4-四氢喹啉-4-醇(7)的合成
在反应瓶中依次加入5150mg的DMF(10mL)溶液,苄溴340mg(2mmol),K2CO3280mg和KI 120mg,搅拌下于80℃反应24h。倾入水中,用乙酸乙酯(3×10mL)萃取,合并萃取液,用无水硫酸钠干燥,浓缩后加入甲醇10mL和NaBH450mg,搅拌下于室温反应30min。加水淬灭反应,用乙酸乙酯(3×10mL)萃取,合并萃取液,用无水硫酸钠干燥,浓缩得黄色固体785mg,产率36%;1H NMRδ:7.32~7.20(m,6H),7.14(d,J=7.5Hz,1H),7.00~7.90(m,1H),6.51(d,J=7.2Hz,1H),6.45(t,J=11.4Hz,1H),5.08(d,J=4.8Hz,1H),4.57(d,J=4.5Hz,1H),4.49(d,J=7.2Hz,2H),3.49~3.48(m,1H),3.32~3.25(m,1H),1.86(t,J=9.3Hz,2H);13C NMRδ: 144.5,138.8,129.2,128.4,128.2,128.0,126.6,126.4,124.8,114.9,110.7,64.2,54.0,44.8,30.3。
(8)1-甲基-1,2,3,4-四氢喹啉-4-醇(8)的合成
在反应瓶中依次加入50.15g,多聚甲醛(PFA)0.6g和NaBH40.19g(5.0mmol),然后立即滴加TFA 1mL,滴毕。搅拌下于室温反应12h。用NaHCO3水溶液调至pH7,用乙酸乙酯萃取,合并萃取液,用无水硫酸钠干燥,浓缩后经硅胶柱层析(洗脱剂:A=1∶2)纯化得红褐色液体865.8mg,产率40%;1H NMRδ:7.14(d,J=7.5Hz,1H),7.09~7.03(m,1H),6.59~6.55(m,2H),5.03(d,J=4.8Hz,1H),4.53(d,J=4.5Hz,1H),3.28~3.21(m,1H),3.14~3.09(m,1H),2.84(s,3H),1.89~1.82(m,2H);13C NMRδ: 146.0,129.1,128.2,125.4,115.4,110.9,64.1,46.1,38.7,30.7。
(9)1,2,3,4-四氢喹啉-4-醇(9)的合成
在反应瓶中依次加入5150mg的甲醇(10mL)溶液和NaBH460mg(1.5mmol),搅拌下于室温反应30min。加水淬灭反应,用乙酸乙酯(3×10mL)萃取,合并萃取液,用无水硫酸钠干燥,减压浓缩后经硅胶柱层析(洗脱剂:A=1∶1)纯化得白色固体90.1g,产率62%;1H NMRδ: 6.43~7.09(m,4H),5.75(s,1H),4.97(d,J=5.1Hz,1H),4.50~4.55(m,1H),3.10~3.39(m,2H),1.68~1.82(m,2H);13C NMRδ: 145.2,129.4,127.8,123.3,114.8,113.4,63.9,36.2,30.4。
2 结果与讨论
在乙酸乙酯/石油醚的展开剂体系中,氮上连有不同保护基团的产物2同底物1比较,表现出不同的相对Rf值。当氮上连有Boc保护基(2a)时,Rf值(0.7)较底物1大,当保护基为乙酰基(2b,0.2)和苯甲酰基(2c,0.3)时,Rf值较1小。
5的合成亦可以通过1的乙酰基保护、氧化得3b;然后在5%NaOH甲醇溶液中于室温脱除乙酰基得5,三步反应总产率为43%。
在8的合成中,如果滴加完TFA,就加入NaHCO3终止反应,此时苄位的羰基仍然存在,再经还原得8。在终止反应调节pH的过程中,pH值调至中性为宜,如果碱性过强,所生成的产物会迅速转变成3,3-二羟基-1-甲基-2,3-二氢喹啉-4(1H)-酮。如果延长反应时间,苄位的羰基在THF-TFA-NaBH4体系中可以还原到羟基,直接一步反应得8。
3 结论
以1,2,3,4-四氢喹啉为原料,经仲胺基团的酰胺化保护、苄位氧化、仲胺脱保护、羰基还原等反应合成了一系列1,2,3,4-四氢喹啉氨基醇类衍生物,建立了一种制备四氢喹啉衍生物的新方法。
该方法一方面为四氢喹啉衍生物合成提供了参考;另一方面,所合成的一系列四氢喹啉醇类化合物可以进行进一步的衍生建立小分子库,用于药物活性分子的筛选。
[1] Nigel B P,John W B,Murray H G M.Disocorhabdin D,an antitumor alkaloid from the sponges latrunculia brevis and prianos sp[J].J Org Chem,1988,53:4127-4128.
[2] Paul D L,Robert W C,Kevin W M,etal.4-amido-2-carboxytetrahydroquinolines.Structure-activity relationships for antagonism at the glycine site of the NMDA receptor[J].J Med Chem,1992,35:1954-1968.
[3] Stephen I A,Geoffrey H,Andrew D Towns.Properties of some novel monoazo disperse dyes derived from ester-substituted tetrahydroquinoline and indoline coupling components[J].Dyes and Pigments,2000,47:33-43.
[4] Raivis Z,Esther H,Andreas P,Christina M.Iridium-catalyzed asymmetric hydrogenation of olefins using pyridine-phosphinites derived from the chiral pool[J].Arkivoc,2008,58-66.
[5] Steven W G,Paul J D.2-substituted 1,2,3,4-tetrahydroquinolines from quinoline[J].Synthesis,1989,221-222.
[6] Kuznetsov V V,Aliev A E,Prostakov N S Prostakov.Synthesis of 2-alkyl(aryl,hetaryl)-4-methyl-12,3,4-tetraahydroquinolines[J].Chem Heterocycl Comp(Engl TRANSL),1994,30:64-68.
[7] Ulrich W,Ralf K,Nikolaus R.Novel iminium compounds fromN-alkylanilinium perchlorates and aldehydes and their unexpected(domino type)reactions[J].J Org Chem,1995,60:2263-2266.
[8] Alan R K,Bogumila R,Stanislaw R.Reactions ofN-alkyl-N-phenyl-1H-benzotriazole-1-methanamines withN-vinylamides andN-vinylcarbazole.A convenient synthesis of 4-(dialkylamino)tetrahydroquinolines[J].J Org Chem,1995,60:3993-4001.
[9] By W S J,Eugene L W,Bennett G B.Cyclization studies in the quinoline series.A new synthesis of 4-aminoquinolines[J].J Am Chem Soc,1949,71:1901-1905.
[10] P Nguyen,E Corpuz,T M. A conveninent synthesis of 7-Halo-1-indanones and 8-Halo-1-tetralones[J].J Org Chem,2003,68:10195-10198.
[11] Gordon W G,Charles F N.Reactions of sodium borohydride in acidic media:N-methylation of amines with paraform aldehyde/trifluoroacetic acid[J].Synth Commun,1987:709-711.
Synthesisof1,2,3,4-Tetrahydroquinolin-4-olDerivatives
CHEN Yong-zheng,ZHENG Dai-jun,LIU Xiao-zu,ZHOU Xiao-jian,XU Ke
(School of Pharmercy,Zunyi Medical University,Zunyi 563000,China)
A series of 1,2,3,4-tetrohydroquinolin-4-ol derivatives with overall yields of 17%~41% were synthesized from 1,2,3,4-tetrahydroquinoline by protection of NH group,oxidation,deprotection and reduction reaction.The structures were confirmed by1H NMR and13C NMR.
tetrahydroquinoline;alkamine;tetrohydroquinolin-4-ol;synthesis
2014-04-09
国家自然科学基金资助项目(21162047,21262051);贵州省科技厅资助项目(黔科合SZ字2012-3078号,黔科合人字2013-047)
陈永正(1982-),男,汉族,贵州遵义人,博士,副教授,主要从事不对称合成的研究。Tel.0852-8609673,E-mail: yzchen@zmc.edu.cn
O626.3
A
1005-1511(2014)05-0687-04
