1H-1,2,4-三唑衍生物爆轰性能的理论计算
2014-08-22马忠亮王建龙李景满
马忠亮,周 华,王建龙,李景满
(1.中北大学化工与环境学院,山西太原030051;2.四川通达化工有限责任公司,四川达州635000)
1H-1,2,4-三唑衍生物爆轰性能的理论计算
马忠亮1,周 华1,王建龙1,李景满2
(1.中北大学化工与环境学院,山西太原030051;2.四川通达化工有限责任公司,四川达州635000)
采用密度泛函理论(DFT)B3LYP方法,在6-31G(d,p)基组水平下对1H-1,2,4-三唑衍生物进行几何构型全优化,设计了合适的等键反应计算1 H-1,2,4-三唑衍生物的生成焓。基于0.001e·bohr-3等电子密度面所包围的体积空间求得分子平均摩尔体积(V)和理论密度(ρ),运用Kamlet-Jacobs方程估算1H-1,2,4-三唑衍生物的爆速(D)和爆压(p),结果表明,1H-1,2,4-三唑发生N-取代或引入-N3基团都能显著提高衍生物的生成焓,-NF2或-ONO2基团能够增强衍生物的爆轰性能。通过计算弱键的化学解离能(BDE),得出部分1H-1,2,4三唑衍生物具有较好的热稳定性。
有机化学;量子化学;密度泛函理论;1H-1,2,4三唑;生成焓;爆轰性能;热稳定性
引 言
低感度、高能量密度化合物是含能材料研究领域的重要方向。三唑是含有3个氮原子的五元环化合物,分子结构中的高氮(50.9%)低碳氢含量不仅使其具有较高的密度,而且更容易达到氧平衡,其衍生物有可能作为高能量密度材料[1]。钝感炸药5-氨基-3-硝基-1,2,4-三唑(ANTA)和3-硝基-1,2,4-三唑-5-酮(NTO)是1,2,4-三唑重要的衍生物,ANTA可用于火箭推进剂,NTO及其盐类是具有广泛应用前景的钝感含能材料。
近年来,三唑衍生物作为含能材料或含能材料中间体备受关注。Krishna kumar等[2]研究了3,5-二硝基-1,2,4-三唑分子间的相互作用;XU L N等[3]研究了NTO分子间的相互作用;Siker A K等[4]研究了ANTA及其衍生物的热稳定性;朱佳平等[5]计算了DNMT的爆轰性能;鲁亚琳[6]研究了1,2,3-三唑及1,2,4-三唑的前线轨道能及热力学性质。为了深入研究取代基团对1 H-1,2,4-三唑性能的影响,本研究对1H-1,2,4-三唑衍生物的爆轰性能进行了系统的计算,以期为1 H-1,2,4-三唑的实验研究提供参考。
1 计算方法
根据取代基种类、位置及数量的不同,将1H-1,2,4-三唑衍生物分为A、B、C、D和E五类(如图1和表1所示)。运用Gaussian03程序[7],采用密度泛函理论(DFT)B3LYP[8-9]方法结合6-31G(d,p)基组,对1 H-1,2,4-三唑衍生物进行分子几何构型优化计算。经振动频率分析,确定优化构型具有势能面上极小点(无虚频),得到分子的总能量。

图1 1H-1,2,4三唑衍生物的分子结构Fig.1 Molecular structure of 1H-1,2,4-triazole derivatives

表1 1H-1,2,4-三唑衍生物的取代基Table 1 Substituent of 1H-1,2,4-triazole derivatives
等键反应广泛用于计算化合物的生成焓[10-11]。本研究设计如下等键反应:
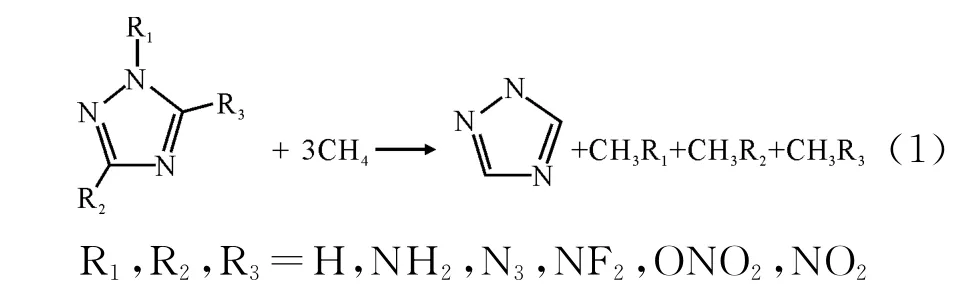
根据式(1)所确定的等键反应计算化合物的气相生成焓,由于含能材料大部分以固相形式存在,在估算爆轰性能时应先计算其固相生成焓。根据盖斯定律:ΔHf,solid=ΔHf,gas-ΔHsub,因此,只需计算升华焓,即可得到目标化合物的固相生成焓。Politzer[12-14]计算升华焓的经验公式为:
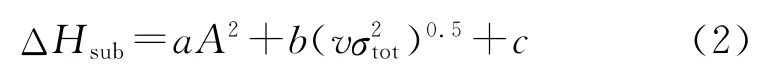
式中:a=11.166×10-4kJ·mol-1A-4;b=6.900 kJ·mol-1;c=12.404 kJ·mol-1[15]
运用Monte-Carlo方法由0.001e/Bohr3等电子密度面所包围的体积求得理论密度,采用公式(3)修正求得其晶体密度:
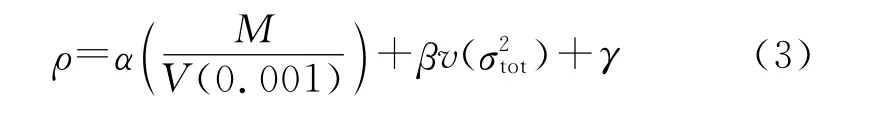
式中:α=0.9183;β=0.0028;γ=0.0443[16]
在求得固相生成焓和晶体密度的基础上,采用Kamlet-Jacobs公式[17-18]估算了1 H-1,2,4-三唑衍生物的爆速和爆压,筛选出爆轰性能较好的化合物。
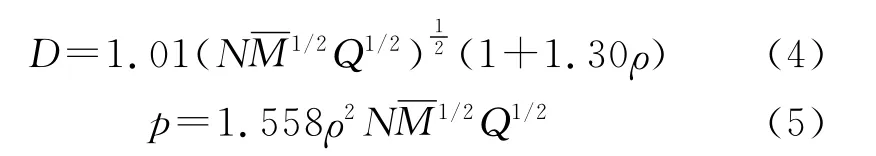
为了考察化合物的热稳定性,以键解离能(BDE)作为衡量化合物热稳定性的一个指标,采用公式(6)计算1 H-1,2,4-三唑衍生物中较弱化学键的键解离能:
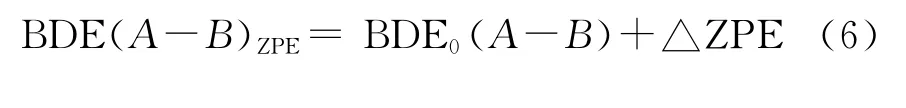
2 结果与讨论
2.1 生成焓
表1列出7种相关化合物的气相生成焓,其中1 H-1,2,4-三唑(C2H3N3)、CH4、CH3NH2、CH3NO2和CH3ONO2生成焓的实验值来自文献[19-21]。由表1可看出,1H-1,2,4-三唑、CH4、CH3NN2、CH3NO2和CH3ONO2的气相生成焓计算值与实验值的相对误差分别为0.39%、3.77%、0.71%、4.32%、8.89%,表明在G2水平下相关化合物的计算值可信。
根据等键反应式(1)计算1 H-1,2,4-三唑衍生物的气相生成焓、升华焓以及固相生成焓,结果见表3。从表3可以看出,1 H-1,2,4-三唑衍生物的生成焓受取代基种类、位置以及数量的影响。观察A、B、C系列可见,发生C-取代时引入-N3基团能大幅度提高衍生物的生成焓,引入-NH2、-NF2或-ONO2基团则导致衍生物的生成焓降低。发生N-取代时(D系列)引入-N3、-NO2、-NH2、-NF2或-ONO2基团都能提高衍生物生成焓,引入基团能够提高衍生物生成焓的顺序为:-N3>-NO2>-NH2>-NF2>-ONO2。
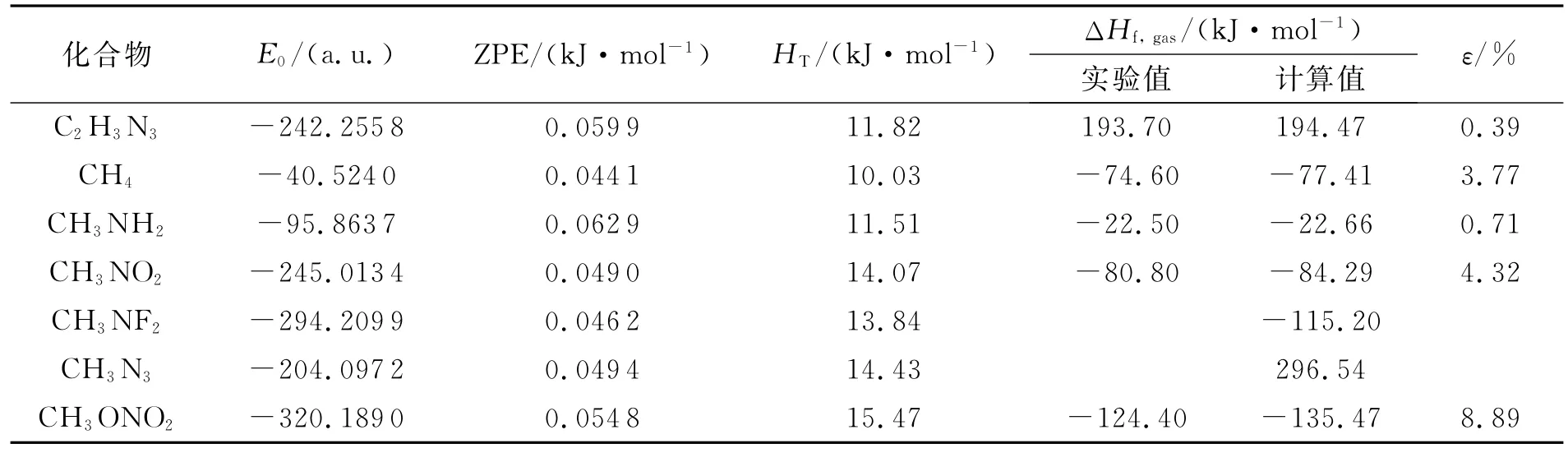
表2 相关化合物的气相生成焓及相对误差Table 2 Heats of formation and relative error for the related compounds
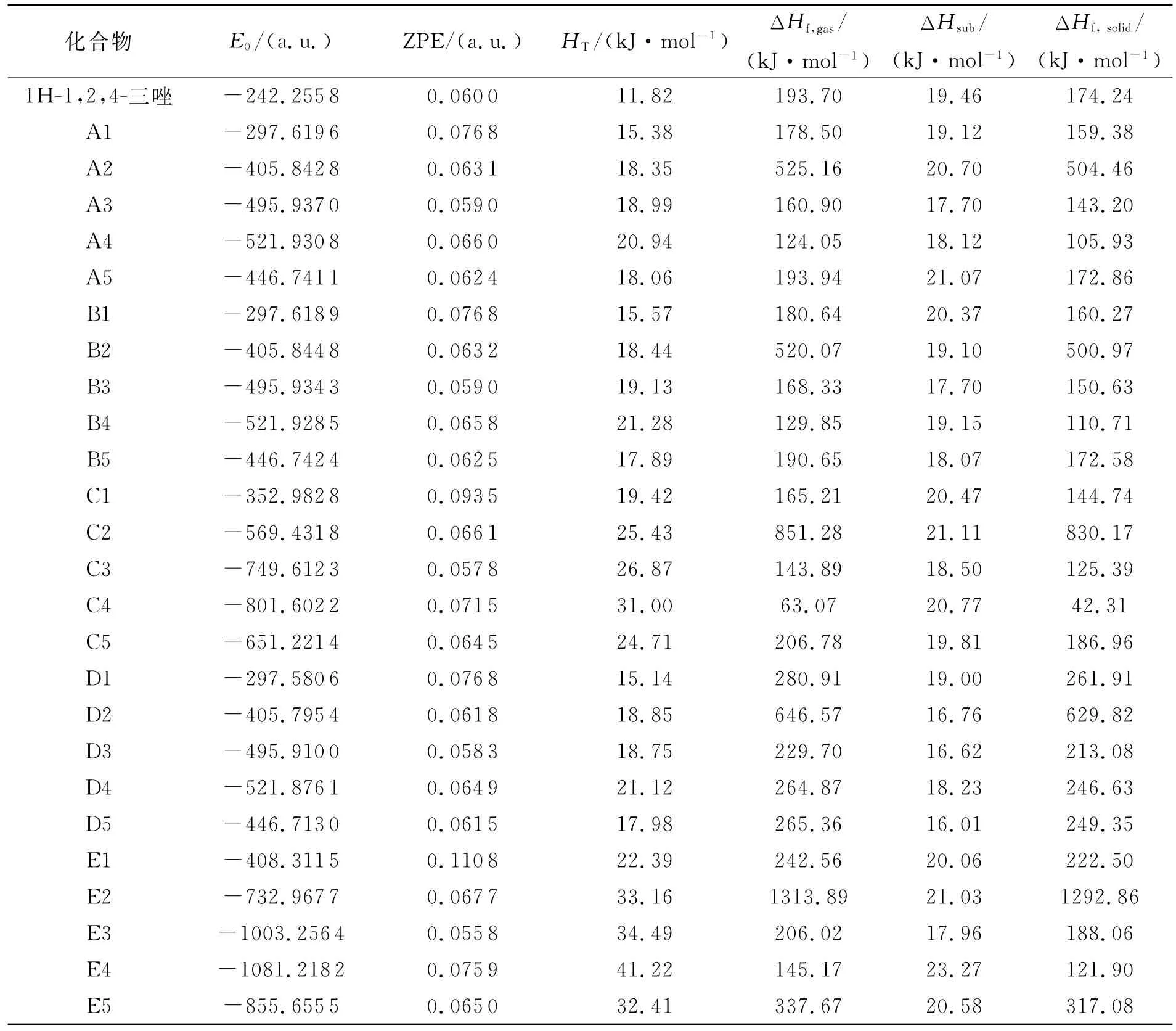
表3 1H-1,2,4-三唑衍生物的总能量、零点能、热校正和生成焓的计算值Table 2 Calculated values of total energies,zero-point energies,thermal correction,heats of formation for the 1H-1,2,4-triazole derivatives
从表3看出,发生C-取代的衍生物,取代基位置对衍生物生成焓的影响并不明显,而发生N-取代的衍生物生成焓则大幅提高。
1 H-1,2,4-三唑衍生物引入基团数与生成焓的关系见表4。从表4可以看出,随着C上的H逐渐被-NH2、-NF2或-ONO2取代,衍生物的生成焓越来越小;而被-N3取代时,衍生物的生成焓随着-N3基团数的增多而增大;当取代基为-NO2时,随着-NO2基团数的增加,衍生物生成焓呈先减小后增大的规律。对-NH2、-N3和-ONO2三种基团衍生物,比较P-Mono(C3)+P-Mono(C5)和P-Di(C,C)的值以及P-Mono(C3)+P-Mono(C5)+PMono(N)和P-Tri(C,C,N)的值,发现1 H-1,2,4-三唑引入-NH2,-N3和-ONO2基团后其衍生物的生成焓有较好的基团加和性。而当取代基为-NF2基团时由于位阻效应,导致衍生物生成焓不符合基团加和性。当引入-NO2基团时,既有位阻效应又能生成弱氢键,同时影响衍生物的生成焓。
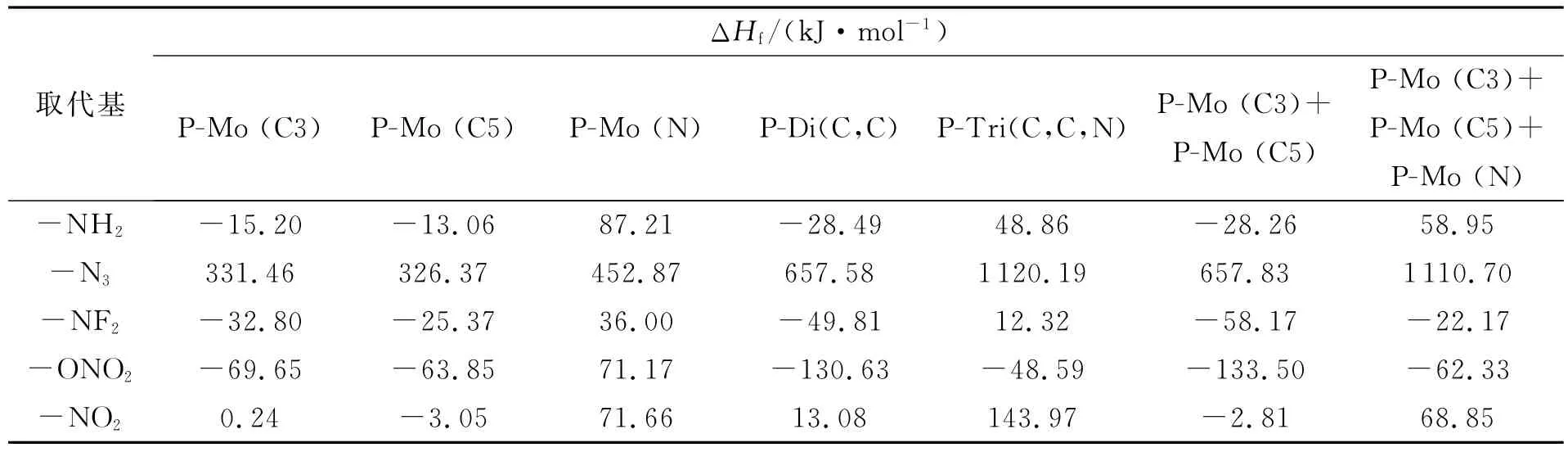
表4 1H-1,2,4-三唑引入取代基数目与生成焓的关系Table 4 Relationship of heats of formation with the substituents number join to 1H-1,2,4-triazole
2.2 能量性能
采用Kamlet-Jacobs公式估算爆速(D)和爆压(p)。从Kamlet-Jacobs公式可以看出,爆速、爆压与密度和爆热有关。运用Monte-Carlo方法由0. 001e/Bohr3等电子密度面所包围的体积求得理论密度,通过修正公式(3)求得其晶体密度,根据得到的固相生成焓计算其爆热。衍生物的密度、爆热、氧平衡、爆速和爆压计算值见表5。
表5可以显示,取代基的种类、位置以及基团数量对衍生物的密度有一定的影响。除A1和D1外,衍生物的密度都大于1 H-1,2,4-三唑的密度,基团对密度增大的贡献顺序为:-NF2>-ONO2>-NO2>-N3>-NH2。同一个基团发生C-取代比发生N-取代得到的衍生物的密度大,而在发生C-取代时取代位置对密度的影响却不大。衍生物密度随着取代基数量的增加而增大,这种增大的趋势随着取代基数量的不断增大而变缓。
从表5可以看出,-NF2、-ONO2、-NO2和-N3基团发生C-取代或N-取代都能提高1H-1,2,4-三唑衍生物的爆热。而-NH2基团发生N-取代时1H-1,2,4-三唑衍生物的爆热增加,但发生C-取代爆热降低。
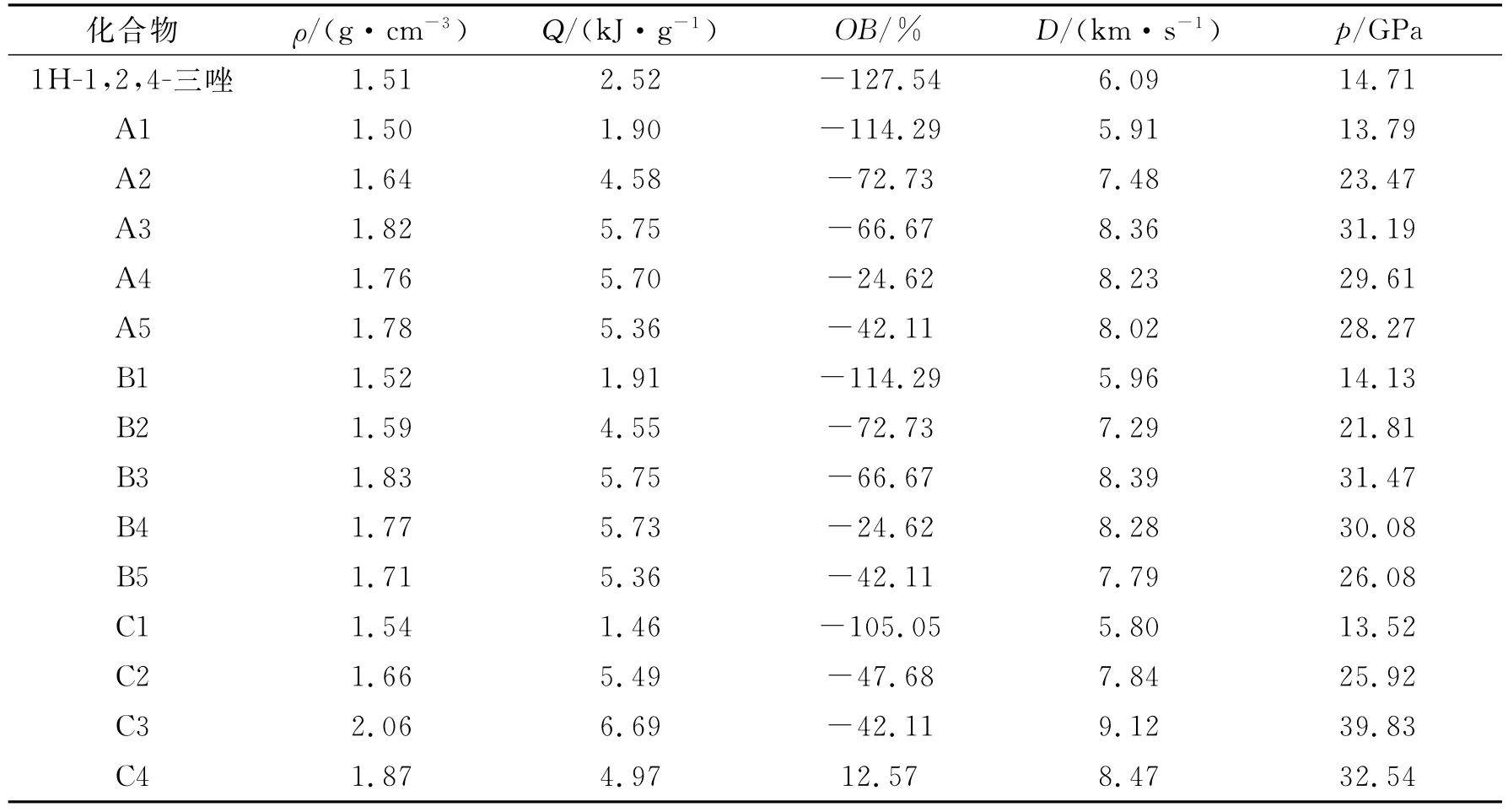
表5 1H-1,2,4-三唑衍生物的密度、爆热、氧平衡、爆速和爆压计算值Table 5 Predicted densities,heats of detonation,oxygen balance,detonation velocities and detonation pressures of 1H-1,2,4-triazole derivatives

续表5
从表5可以看出,密度和爆热受取代基团类型、取代位置和取代数量的影响而变化,根据Kamlet-Jacobs公式,密度和爆热的变化决定了爆速和爆压的变化。因此,爆速和爆压也与取代基的类型、取代位置和取代数量有关。-NF2、-NO2、-NO2和-N3基团不但能提高衍生物的密度,同时还能提高其爆热,因此这些基团的引入可大幅度提高1 H-1,2,-4-三唑衍生物的爆轰性能。
由表5可以看出,C3,C5,E3和E5的爆轰性能高于RDX,但是只有C3和E3的爆轰性能优于HMX。
2.3 热稳定性
在计算过程中根据Mulliken键级选择相对较弱的键,由式(6)计算其化学键解离能(BDE)。表6列出1 H-1,2,4-三唑衍生物中弱键的解离能和Mulliken键级。对于发生C-取代时(A、B和C系列),含-NH2取代基时环内NI-N2键的BDE小于C-R键,说明衍生物开环反应是诱导反应;相反,含-NO2和-N3基团的衍生物环内N1-N2键的BDE大于C3-R键,说明在衍生物中取代基的脱落是诱导反应;含-NF2和-ONO2基团的衍生物,N-F和O-NO2键的断裂是解离过程中最容易发生的。而发生N-取代(D和E系列),并且只有-ONO2基团引入到N1位置时,O-NO2键最弱;当-NH2、-N3、-NF2或-NO2引入到N1位置时,N1-R键的BDE远小于其他键,说明1H-1,2,4-三唑衍生物中只要发生N-取代,化合物中最可能发生断裂的键是N1-R键,而-ONO2基团引入到N1位置时,最容易发生断裂的键则是ONO2键。
Chung[23]和Su[24]等提出,作为新的高能材料候选物的首要条件是其键解离能应超过80k J/mol。从表5数据可知,所有目标化合物的键解离能高于80 kJ/mol,但在实际应用中有待进一步实验验证。
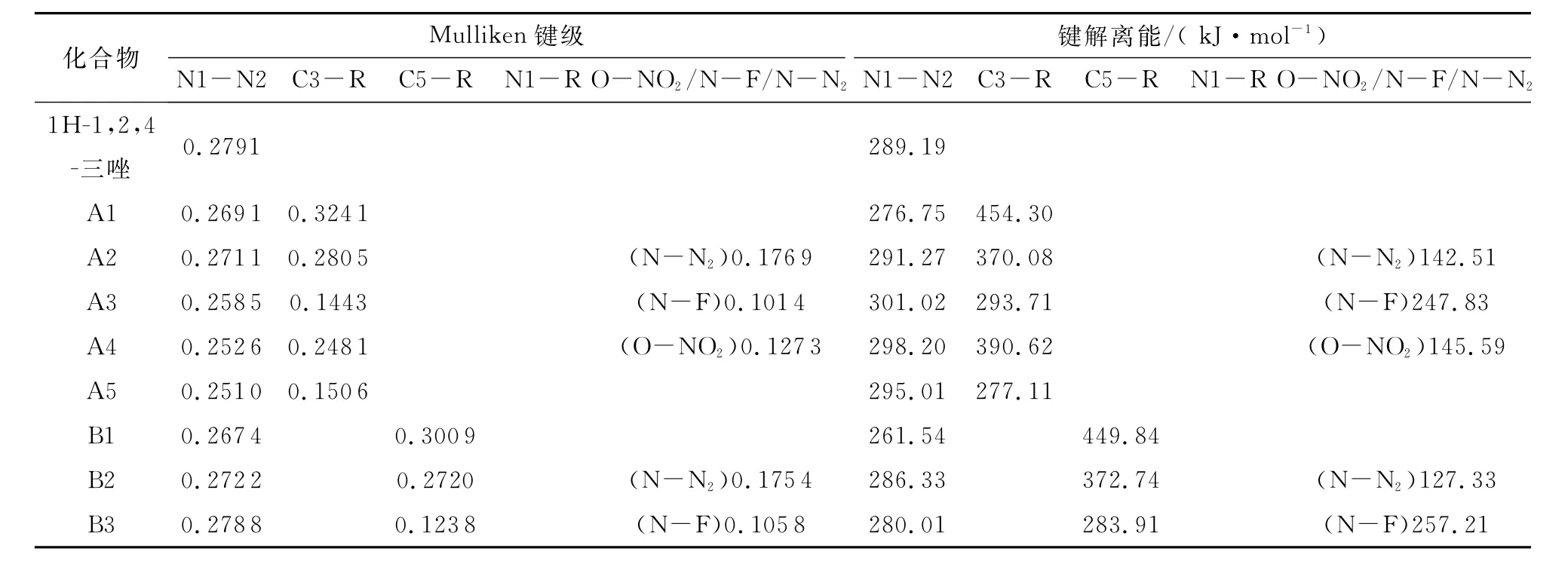
表6 1H-1,2,4-三唑衍生物中弱键的解离能和Mulliken键级Table 6 Bond dissociation energies and Mulliken bond orders of the relatively weak bonds in the 1H-1,2,4-triazole derivatives
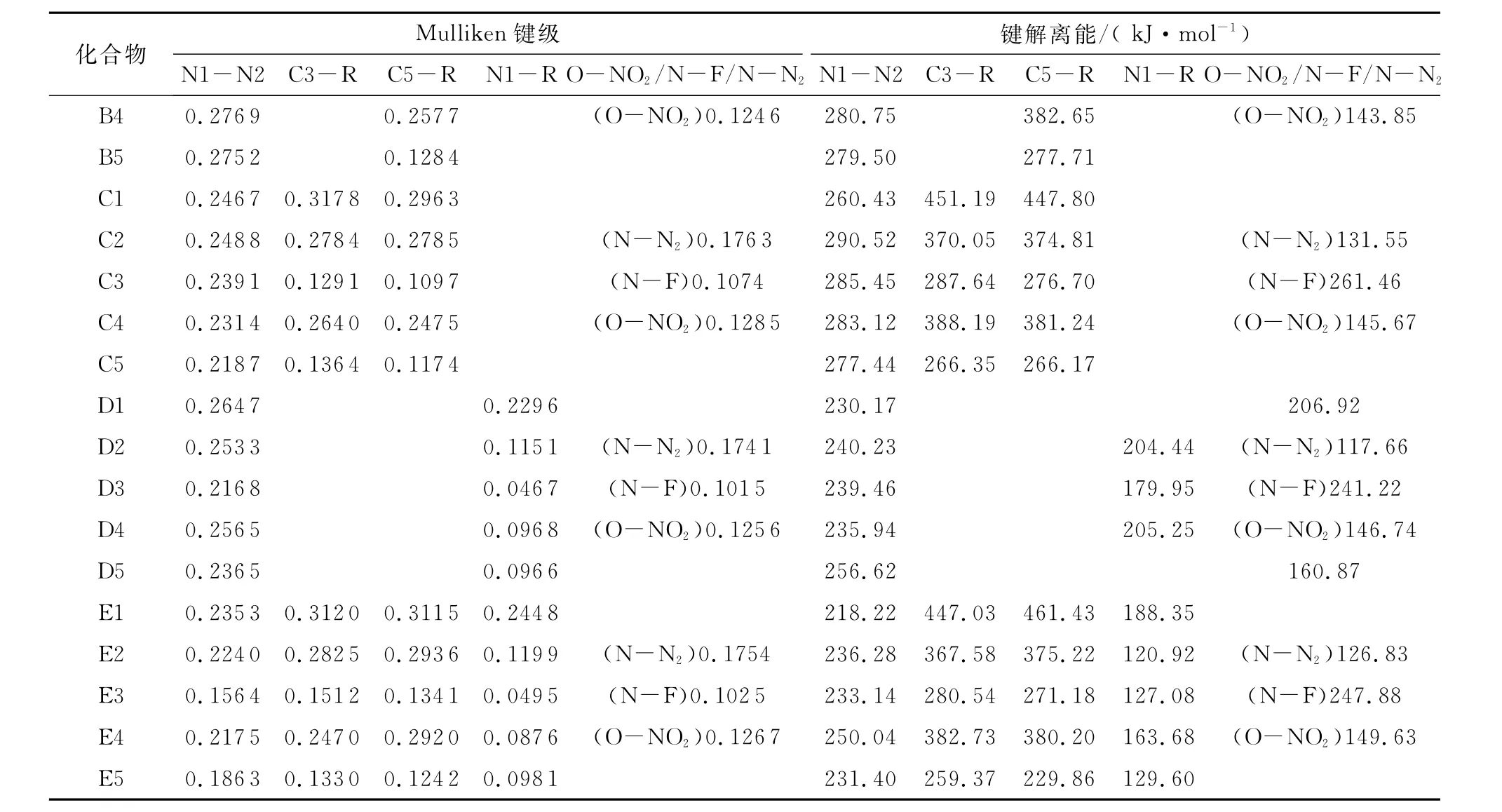
续表6
3 结 论
(1)运用DFT-B3LYP方法研究了1H-1,2,4-三唑衍生物的生成焓、爆轰性能和热稳定性。取代基的种类和位置影响衍生物的生成焓,引入-N3基团能显著增加衍生物的生成焓。对于同类取代基发生N-取代衍生物生成焓值高于发生C-取代衍生物。
(2)-NF2、-ONO2、-NO2和-N3基团不仅能提高衍生物的密度,还能提高其爆热,有效提高1H-1,2,4-三唑衍生物的爆轰性能。所有衍生物的热稳定性较好。综合考虑爆轰性能和热稳定性,C3、C5、E3和E5有可能成为潜在的高能量密度材料。
[1] 阳世清,岳守体.国外四嗪四唑类高氮含能材料研究进展[J].含能材料,2003,11(4):231-235.
YANG Shi-qing,YUE Shou-ti.Progress in high-nitrogen energetic materials derails derived from tetrazine and tetrazole[J].Chinese Journal of Energetic Materials,2003,11(4):231-235.
[2] Krishna K V,Keresztury G,Sundius T,et al.Hydrogen bonding and molecular vibrations of 3,5-diamino-1,2,4-triazole[J].Spectrochim Acta A,2005,61:261-267.
[3] Xu L N,Xiao H M,Fang G Y,et al.Theoretical study on intermolecular interactions of 3-nitro-1,2, 4triazole-5-one-dimers[J].Acta Chimica Sinica,2005,63(12):1062-1068.
[4] Sikder A K,Geetha M,Sarwade D B,et al.Studies on characterization and thermal behaviour of 3-amino-5-nitro-1,2,4-triazole and its derivatives[J].Hazard,2001,82(1):1-12.
[5] 朱佳平,刘胜楠,李永祥.几种唑类炸药分子的结构和爆轰性能的密度泛函理论研究[J].中北大学学报(自然科学版),2011,2:200-206.
ZHU Jia-ping,LIU Sheng-nan,LI Yong-xiang. Structures and detonation performance for several explosive molecule of thiiazoles by density functional theory[J].Chinese Journal of North University of China(Natural Science Edition),2011,2:200-206.
[6] Lu Y L,Gong X D,Ju X H.structures and properties of 1,2,3-triazoles and 1,2,4-triazoles[J].Chinese J Struct Chem,2006,25(5):582-588.
[7] Frisch M J,Trucks G W,Schlegel H B.et al.Gaussian 03,Revision B.03.[M].Pittsburgh:Gaussian Inc,2003.
[8] Lee C,Yang W,Parr R G.Development of the collsalvetti correlation-energy formula into a functional of the electron density[J].Phys Rev B,1988,37:785-789.
[9] Becke A D.Density-functional thermochemistry.Ⅲ. the role of exact exchange[J].Chem Phys,1993,98:5648-5652.
[10]Zhang X W,Zhu W H,Xiao H M.Comparative theoretical studies of energetic substituted carbon-and ni-trogen-bridged difurazans[J].Phys Chem A,2010,114:603-612.
[11]Ju X H,Wang X,Bei F L.Substituent effects on heats of formation,group interactions,and detonation properties of polyazidocubanes[J].Comput Chem,2005,26(12):1263-1269.
[12]Politzer P,Lane P,Murray J S.Computational characterization of a potential energetic compound:1,3,5,7-Tetranitro-2,4,6,8-tetraazacubane[J].Cent Eur J Energ Mater,2011,8:39-52.
[13]Politzer P,Murray J S.Some perspectives on estimating detonation properties of C,H,N,O compounds[J].Cent Eur J Energ Mater,2011,8:209-220.
[14]Politzer P,Murray J S,Grice M E,et al.Calculation of heats of sublimation and solid phase heats of formation[J].Mol Phys,1997,91:923-928.
[15]Byrd E F C,Rice B M.Improved prediction of heats of formation of energetic materials using quantum chemical calculations[J].Phys Chem A,2006,110:1005-1013.
[16]Politzer P,Martinez J,Murray JS.et al.An electrostatic interaction correction for improved crystal density predictions[J].Mol Phys,2009,107:2095-2101.
[17]Kamlet M J,Jacobs S J.Chemistry of detonation.I.A simple method for calculating detonation properties of C,H,N,O explosives[J].Chem Phys,1968,48:23-35.
[18]Kamlet M J,Ablard J E.Chemistry of detonations. II.buffered equilibria[J].Chem Phys,1968,48:36-42.
[19]Dean J A.Lange′s Handbook of Chemistry[M].15th Ed.New York:McGraw-Hill,1999.
[20]David R L.Handbook of chemistry and physics[M]. 84th edn.Boca Raton:CRC Press,2003-2004.
[21]Afeefy H Y,Liebman J F,Stein S E.Neutral Thermochemical Data[M].Gaithersburg:National Institute of Standards and Technology,2000.
[22]Talawar M B,Sivabalan R,Mukundan T,et al.Environmentally compatible next generation green energetic materials(GEMs)[J].Hazard Mater,2009,161:589-607.
[23]Chung G S,Schimidt M W,Gordon M S.An ab initio study of potential energy surfaces for N8 isomers[J]. Phys Chem A,2000,104:5647-5650.
[24]Su X F,Cheng X L,Meng C M.et al.Quantum chemical study on nitroimidazole,polynitroimidazole and their methyl derivatives[J].Hazard Mater,2009,161(1):551-558.
Theoretical Calculation on Detonation Performance of 1H-1,2,4-Triazole Derivatives
MA Zhong-liang1,ZHOU Hua1,WANG Jian-long1,LI Jing-man2
(1.School of Chemical Engineering and Environment,North University of China,Taiyuan 030051,China;2.Sichuan Tongda Chemical Industry Co.,Ltd,Dazhou Sichuan 635000,China)
The geometries of 1H-1,2,4-triazole derivatives were obtained by density functional theory at DFTB3LYP/6-31G(d,p)level.The heats of formation(HOF)were predicted by designed isodesmic reactions.The average molar volume(V)and theoretical density(ρ)were estimated based on 0.001 e·bohr-3density space.The detonation velocity(D)and pressure(p)of 1H-1,2,4-triazole derivatives were estimated by using the Kamlet-Jacbos equation.It was found that N-substituent in 1H-1,2,4-triazole can improve the HOF values of the derivatives,and-N3is an effective structural unit for improving the HOF values of the derivatives.The-NF2or-ONO2group is very useful for enhancing the detonation performance of these derivatives.The analysis of the bond dissociation energies for several relatively weak bonds suggest that most of the derivatives have good thermal stability.
organic chemistry;quantum chemistry;density functional theory;1H-1,2,4-triazole derivatives;heat of formation;detonation performance;thermal stability
TJ55;O<641 文献标志码:A class="emphasis_bold">641 文献标志码:A 文章编号:1007-7812(2014)04-0038-07641 文献标志码:A
1007-7812(2014)04-0038-07
A 文章编号:1007-7812(2014)04-0038-07
2014-03-10;
2014-05-30
马忠亮(1967-),男,教授,博导,从事新型发射药及装药研究。
