脂多糖加速饮食所致非酒精性脂肪肝小鼠的炎症和氧化应激
2014-08-15史国兵张敬一
张 威,史国兵,张敬一
0 引言
非酒精性脂肪肝(Nonalcoholic fatty liver disease,NAFLD)是一种最常见的慢性肝脏疾病,在成人和儿童中都有较高的发病率[1],非酒精性脂肪肝炎(Non-alcoholic steatohepatitis,NASH)的特点是具有显著的脂肪病变、炎症和进行性的肝纤维化,最终导致肝硬化甚至是肝癌的发生[2]。肠道细菌、细菌毒素以及内毒素诱导的前炎症因子(如肿瘤坏死因子-α能激起脂肪性肝炎的坏死性炎症病变和肝纤维化[3-4],并且有临床研究表明,NASH患者肠道中的菌群含量显著高于正常受试者[5]。另外,有临床以及实验室研究证明抗菌药物能够改善肝功能[6-7]。提示肠道来源的细菌性内毒素可能会诱发NASH的发生,因此,本研究旨在探讨脂多糖在饮食诱导的NASH模型中的作用。
1 材料和方法
1.1 实验动物及分组 本实验选用8~10周龄雄性C57BL/6小鼠,体重(20±2) g,购自中国医科大学动物实验中心,动物合格证号:SCXK(辽)2008-0005。实验动物常规饲养于沈阳军区总医院清洁级动物房,12 h光照和黑夜循环,温度(22±2)℃,湿度50%~60%。MCD饲料的配方参考国外文献[8]。MCS饲料配方[9],即MCD对照饲料,是在MCD饲料配方基础上,加上胆碱2 g/kg、蛋氨酸3 g/kg。MCD及MCS饲料均由江苏南通特洛菲饲料有限公司加工制作,为清洁级饲料,4 ℃低温保存。动物分为3组,MCS或MCD饲料喂养2周,每周2次腹腔注射生理盐水或脂多糖(LPS),组别如下:MCS+Saline组给予MCS+生理盐水腹腔注射,MCD+Saline组给予MCD+生理盐水腹腔注射,MCD+LPS组给予MCD+1 mg/kg LPS腹腔注射(购自Sigma)。
1.2 标本处理及组织病理学检测 各组小鼠末次腹腔注射6 h后乙醚麻醉,摘取一侧眼球后取血,3 000 rpm离心后留上清冻存备用。脱颈处死动物取部分肝小叶用10%福尔马林固定,石蜡包埋,进行HE染色和Sirius Red染色,另取部分肝脏于-80 ℃冻存待测。
1.3 血清ALT、AST和TNF-α含量测定 ALT、AST按照试剂盒要求采用全自动生化分析仪进行测定,检测TNF-α的ELISA试剂盒购自R&D公司,按试剂盒的操作规程对上述指标进行检测。
1.4 肝脏TBARS含量测定 TBARS(硫代巴比妥酸反应物)能够反映氧化应激水平,按照LabAssayTM的TBARS试剂盒的操作规程检测各组小鼠肝脏中的TBARS水平。
2 结果
2.1 脂多糖加速NASH小鼠的肝脏炎性发生 肝脏组织HE染色可见:MCS+Saline组小鼠肝组织肝窦清晰可见,肝索排列整齐,形态结构均正常;MCD+Saline组小鼠肝组织充满大量大小不一的脂肪空泡,肝细胞结构紊乱呈气球样变,有部分炎性细胞浸润明显,呈灶状分布,炎性细胞以单核细胞、淋巴细胞为主;MCD+LPS组同样也呈现出脂肪肝炎的特点,但炎性细胞浸润较多,局部呈现坏死状病变。见图1。
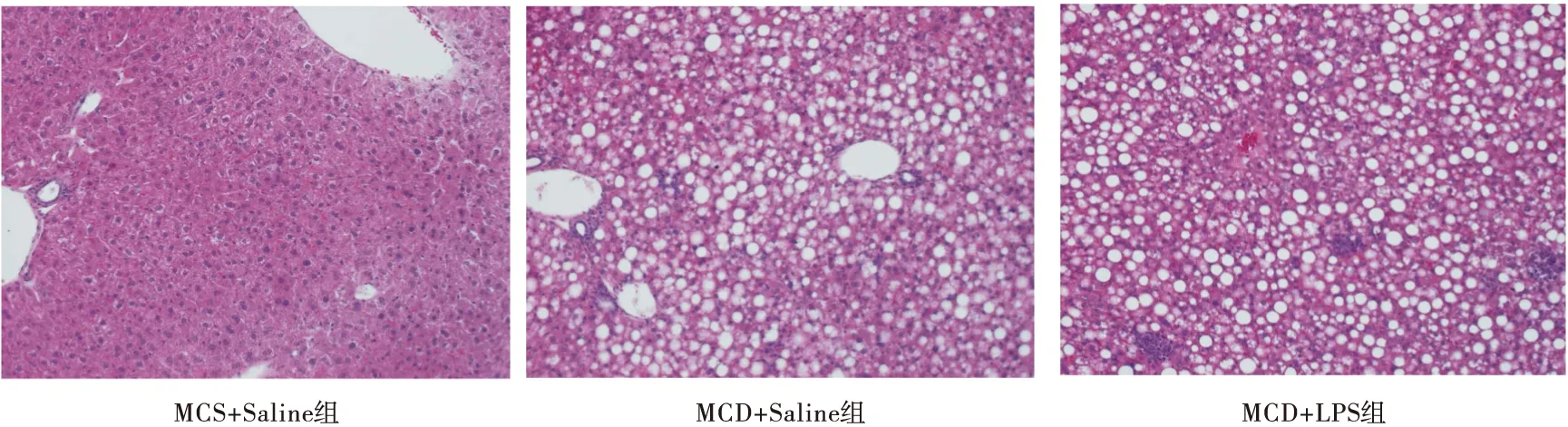
图1 小鼠肝脏HE染色(×200)
2.2 脂多糖加速NASH小鼠的肝纤维化 通过Sirius Red染色观察小鼠肝脏的纤维化变化(见图2)。结果发现,MCS+Saline组除血管外,无明显红色着色现象;MCD+Saline组的肝脏组织内可见轻度的纤维化发生;MCD+LPS组的肝脏组织内则可见较多的纤维化发生。

图2 小鼠肝脏Sirius red染色(×200)
2.3 脂多糖加速NASH小鼠的肝功能受损 血清ALT、AST指标检测结果见图3。由图3可见,与MCS组相比,MCD两组的血清ALT和AST都显著升高(P<0.01),另外,腹腔注射LPS进一步使肝功能受损,AST和ALT的含量显著高于生理盐水注射组(P<0.05)。
2.4 脂多糖促进NASH小鼠血清中TNF-α的表达 用ELISA试剂盒对血清中TNF-α的含量进行检测(图4),结果发现MCD饮食可导致血清TNF-α含量轻度增加(P<0.05),但当注射LPS后,血清中的TNF-α含量增加了约10倍(P<0.01)。
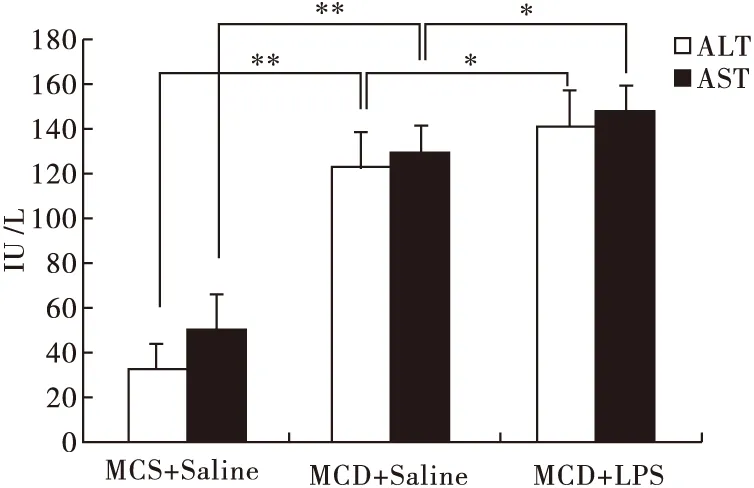
图3 LPS对NASH小鼠血清ALT和AST的影响

图4 LPS对NASH小鼠血清TNF-α的影响
2.5 脂多糖进一步加重NASH小鼠肝脏的氧化应激 对肝脏中的TBARS含量进行检测发现(图5):与MCS正常饮食组相比,MCD饮食组可造成小鼠肝脏氧化应激水平显著升高(P<0.01),腹腔注射LPS后进一步加重了肝脏的氧化应激水平(P<0.01)。
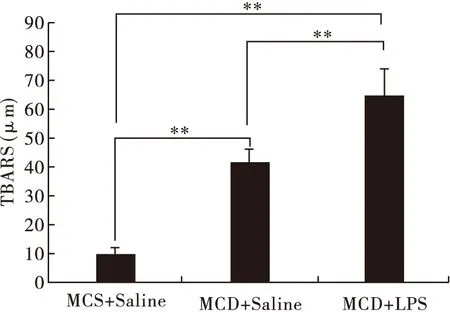
图5 LPS对NASH小鼠肝脏TBARS的影响
3 讨论
近年来研究发现,肠道菌群的移位和紊乱与患者发生慢性肝脏疾病和肝硬化有重要关系[10]。在几种类型的慢性肝病中,均发现血液中LPS的含量升高。肠道上皮的完整性受损导致其对细菌的通透性改变,因此,肠道细菌发生移位,进入腹腔,之后细菌代谢产生的脂多糖等有害物质加速了肝脏疾病的发展[11]。
本实验旨在探讨LPS对NASH发病进展的研究,因此,我们选用了MCD饮食诱导的NASH小鼠模型。该模型是国际上公认的用于研究NAFLD中炎症及纤维化的最好的动物模型之一[12]。NASH是NAFLD向非酒精性脂肪性肝硬化(NAC)进展过程中的重要环节,是隐源性肝硬化的重要原因之一[2]。NASH发病机制不明确,目前国际上公认的为“二次打击学说”[13-14]。肝脏的脂肪变性作为“第一次打击”,易化肝细胞对其他作为“二次打击”的因素的敏感性,其中可作为“二次打击”的物质有很多,包括药物、超氧阴离子、细胞因子等,这些因素最终导致肝细胞的凋亡及炎性细胞的产生。另有研究发现,NASH患者的肝细胞凋亡程度与病情呈正相关[15-16]。肝细胞凋亡能够激活肝星状细胞,进而使之释放细胞外基质[17-18],这些基质的沉积是引起纤维化发生的根本原因。
本实验结果显示,给予小鼠2周的MCD饮食可见明显的NAFLD症状,肝脏内有大量不规则脂肪滴形成,肝组织结构紊乱并出现炎性细胞浸润,AST和ALT表达增加;当腹腔注射LPS后,炎症情况增加,并出现坏死前兆,肝纤维化增加,AST和ALT含量进一步增加,肝功能受损加重,并且血清TNF-α含量增加,肝脏氧化应激加重。LPS能够作用于Kupffer细胞上的CD14和Toll样受体-4,从而引起TNF-α的合成增多[19]。TNF-α是一种“二次打击”的因子,能够使细胞产生氧化应激,使干细胞内产生大量的活性氧集团(ROS),从而进一步导致细胞凋亡[20]。
综上所述,本实验在MCD饮食所致NASH小鼠模型的基础上腹腔注射LPS,引起NASH的病程进展,血清中的TNF-α增加,肝脏氧化应激增加,纤维化增加引起肝功能进一步受损,病程出现不可逆性进展。本研究具有转化医学特色,结合临床发现部分肝病患者服用抗生素后肝功能指标好转,提示肠道菌群及其释放的内毒素是加重肝病进展的重要原因之一,希望通过今后更加深入的研究,特别是信号通路方面的进一步探索,为临床提供治疗依据,为患者提供更好的治疗方案。
参考文献:
[1] Roberts EA.Nonalcoholic steatohepatitis in children[J].Curr Gastroenterol Rep,2003,5(3):253-259.
[2] Powell EE,Cooksley WGE,Hanson R,et al.The natural history of nonalcoholic steatohepatitis:a follow-up study of 42 patients for up to 21 years [J].Hepatology,1990,11(1):74-80.
[3] Iimuro Y,Gallucci RM,Luster MI,et al.Antibodies to tumor necrosis factor alfa attenuate hepatic necrosis and inflammation caused by chronic exposure to ethanol in the rat[J].Hepatology,1997,26(6):1530-1537.
[4] Adachi Y,Moore LE,Bradford BU,et al.Antibiotics prevent liver injury in rats following long-term exposure to ethanol[J].Gastroenterology,1995,108(1):218-224.
[5] Wigg AJ,Roberts-Thomson IC,Dymock RB,et al.The role of small intestinal bacterial overgrowth,intestinal permeability,endotoxaemia,and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis[J].Gut,2001,48(2):206-211.
[6] Li Z,Yang S,Lin H,et al.Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease[J].Hepatology,2003,37(2):343-350.
[7] Loguercio C,De Simone T,Federico A,et al.Gut-liver axis:a new point of attack to treat chronic liver damage[J].Am J Gastroenterol,2002,97(8):2144-2146.
[8] Lee CS,Yan JS,Ng RK,et a1.Polyunsanturated fat in the methinine-choline deficient diet influences hepatic inflammation but not hepatocellular injury [J].J Lipid Res,2007,48 (8):1885-1896.
[9] Fizwm E,Loewno A,Bianca G,et a1.Mice fed a lipogenic methionine choline-deficient diet develop hypermetabolism coincident with hepatic suppression of SCD-1[J].J Lipid Res,2006,47(10):2280-2290.
[10]Almeida J,Galhenage S,Yu J,et al.Gut flora and bacterial translocation in chronic liver disease[J].World J Gastroenterol,2006,12(10):1493-1502.
[11] Ilan Y.Leaky gut and the liver:a role for bacterial translocation in nonalcoholic steatohepatitis[J].World J Gastroenterol,2012,18(21):2609-2618.
[12] Su GL.Lipopolysaccharides in liver injury:molecular mechanisms of Kupffer cell activation[J].Am J Physiol Gastrointest Liver Physiol,2002,283(2):256-265.
[13] Basaranoglu M,Basaranoglu G,Sentürk H.From fatty liver to fibrosis:a tale of “second hit”[J].World J Gastroenterol,2013,19(8):1158-1165.
[14]陈兴梅.非酒精性脂肪肝患者肝损伤与代谢异常及C反应蛋白的关系[J].中国医药,2012,7(4):504.
[15]Wieckowska A,Zein NN,Yerian LM,et a1.In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease[J].Hepatology,2006,44(1):27-33.
[16] Feldstein AE,Canbay A,Angulo P,et a1.Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis[J].Gastroenterology,2003,125(2):437-443.
[17] Kahraman A,Barreyro FJ,Bronk SF,et al.TRAIL mediates liver injury by the innate immune system in the bile duct-ligated mouse[J].Hepatology,2008,47(4):1317-1330.
[18] Canbay A,Friedman S,Gores GJ.Apoptosis:the nexus of liver injury and fibrosis[J].Hepatology,2004,39(2):273-278.
[19]Anstee QM,Goldin RD.Mouse models in non-alcoholic fatty liver disease and steatohepatitis research[J].Int J Exp Pathol,2006,87(1):1-16.
[20] Wei Y,Rector RS,Thyfault JP,et al.Nonalcoholic fatty liver disease and mitochondrial dysfunction[J].World J Gastroenterol,2008,14(2):193-199.
