神经氨酸酶抑制剂的研究进展
2014-08-14张建国张继宁黄勋娟
张建国,张继宁,黄勋娟
(1.上海理工大学医疗器械与食品学院,上海200093;2.同济大学环境科学与工程学院,上海200092)
神经氨酸酶是水解唾液酸(N-乙酰-神经氨酸)与糖蛋白之间糖苷键的一种水解酶。根据糖苷键位置的不同可以将神经氨酸酶分为外切酶(E3.2.1.18)和内切酶(E3.2.1.129)。神经氨酸酶广泛存在于各种微生物中,并有助于微生物在动物体表生存。1947年,神经氨酸酶首先在纯培养的霍乱弧菌和产气荚膜梭菌中被发现[1],随后,在其它70多种微生物和病毒中发现了神经氨酸酶;同时,在原生动物和非致病性的微生物中也发现了神经氨酸酶。同一种生物中有多种神经氨酸酶,例如粘性放线菌、脆弱拟杆菌、气肿疽梭菌等中有水解α(2,3)-、α(2,6)-、α(2,8)-键的多种神经氨酸酶[2]。哺乳动物中有4种神经氨酸酶 NEU1、NEU2、NEU3、NEU4[3],随机分布于生物的细胞器中:NEU1、NEU2、NEU3分别存在于溶酶体、细胞质和细胞质膜上;NEU4存在于溶酶体、线粒体和细胞内膜多个细胞器上。
早在1957年就发现病毒具有水解糖蛋白上糖苷键的能力[4]。由于研究手段的限制,1983年才确定神经氨酸酶的晶体结构[5],随后研究人员利用神经氨酸酶和神经氨酸的复合晶体结构确定了神经氨酸酶的作用机理[6]:神经氨酸酶在水解过程中和糖蛋白形成一个神经氨酸酶阳离子的过渡态,水分子介入后从糖蛋白中释放出唾液酸[7]。神经氨酸酶在病毒侵染宿主过程中有两方面的作用:(1)病毒靠近宿主细胞时,病毒表面的神经氨酸酶水解掉宿主细胞表面糖蛋白上的唾液酸残基,有利于病毒核进入宿主细胞内;(2)子代新病毒利用神经氨酸酶和神经氨酸的相互作用,粘附在宿主细胞表面,等待扩散到周围的宿主细胞[8]。
1 神经氨酸酶的结构
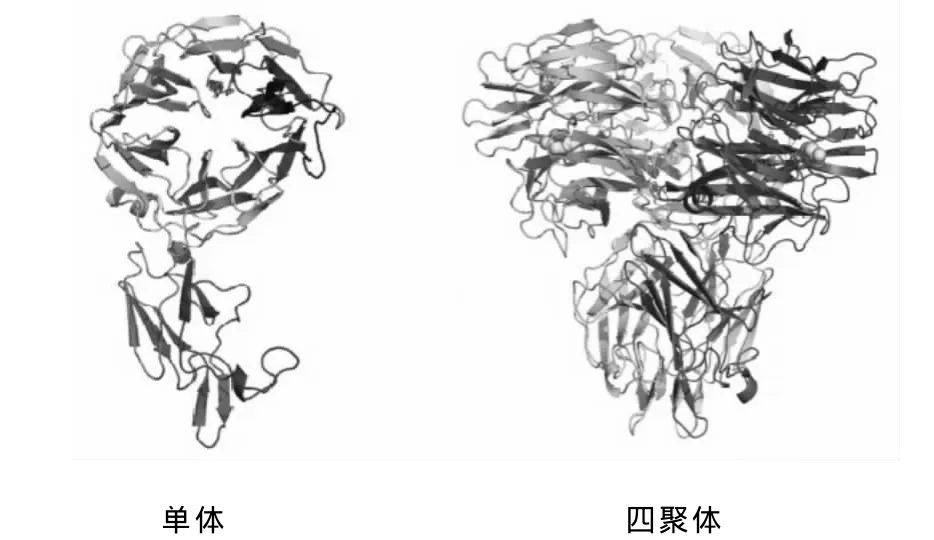
图1 铜绿假单胞菌神经氨酸酶的结构Fig.1 Structure of neuraminidase fromPseudomonas aeruginosa
神经氨酸酶的结构是四聚体,每个单体都含有球形的头部和细长的颈部(图1)[9]。流感病毒可以分为A、B、C 3个亚型[10]。神经氨酸酶以颈部固定在病毒的表面,以球形的头部水解糖蛋白的神经氨酸酶活性部位。分析发现,神经氨酸酶的活性中心是严格的保守区域,由10个极性氨基酸残基和4个疏水氨基酸残基组成,含有6个逆时针方向排列的β折叠片。图2是神经氨酸酶活性中心的示意图[11]。

图2 神经氨酸酶活性中心的示意图Fig.2 The schematic diagram of neuraminidase active center
2 神经氨酸酶的应用
神经氨酸酶的主要功能是帮助微生物或病毒水解糖蛋白的唾液酸残基,被广泛应用于临床疾病的诊断[2],如治疗因神经氨酸酶缺失造成的笨拙综合症[1]。神经氨酸酶水解神经节苷酯的唾液酸残基,形成含有不同分子数唾液酸的神经节苷酯。其中,单唾液酸四己糖神经节苷酯(GM1)是有效治疗脑神经损伤的药物,目前已得到广泛应用。
由于化学法制备GM1存在着制备效率低、产品不安全等不足,所以找到高效、安全的GM1制备方法就尤为重要。Peng等[12]报道了一种来源于乳酪短杆菌的神经氨酸酶具有优良的性质:专一性水解神经节苷酯生产GM1,效率高,水解条件温合,过程简单。Zhang等[13]将乳酪短杆菌神经氨酸酶基因在大肠杆菌中表达后得到更高的酶活力,为大量制备神经氨酸酶和GM1奠定了基础。由于神经氨酸酶在病毒侵染宿主过程中的重要作用和神经氨酸酶活性中心保守的特征,神经氨酸酶成为神经氨酸酶抑制剂的主要靶点。
3 神经氨酸酶抑制剂的研究
3.1 神经氨酸类似物
由于神经氨酸酶活性中心是严格的保守结构,据此设计1个与神经氨酸酶活性中心结合的化学分子就是神经氨酸酶抑制剂研究的总体思路[14]。神经氨酸酶活性中心和神经氨酸分子的相互作用已经被解析[6],唾液酸是设计新型神经氨酸酶抑制剂分子的天然模板。2-脱氧-α-唾液酸及其衍生物是最先在病毒神经氨酸酶晶体结构解析后根据其活性中心分子作用设计的抑制剂。2-脱氧-2,3-双脱氢-唾液酸(Neu5Ac2en)是经过结构改进之后经体内试验证实比较有效的1个抑制剂[15],也是后续新抑制剂的设计模板[8]。经过计算机辅助软件GRID进一步改进,将Neu5Ac2en的4位羟基替换为氨基或胍基后更契合神经氨酸酶的活性中心,具有更好的抑制效果[16-17],其中4-氨基-4-脱氧 Neu5Ac2en的抑制效果比 Neu5Ac2en的抑制效果高100倍[8]、4-胍基-4-脱氧Neu5Ac2en的抑制效果比Neu5Ac2en的抑制效果高10 000倍[18]。4-胍基-4-脱氧 Neu5Ac2en的抑制效果比4-氨基-4-脱氧Neu5Ac2en的抑制效果更好是由于它除了和Glu119发生相互作用外,还和Glu229发生相互作用;此外,这两种抑制剂对A型和B型病毒均有抑制效果[16]。4-胍基-4-脱氧 Neu5Ac2en被葛兰素史克命名为zanamivir,zanamivir是第一个被FDA批准销售的抗病毒药物,目前在市场上作为吸入式喷雾剂销售,近年来的研究发现,神经氨酸酶通过变异对zanamivir产生耐药性[19]。研究表明,唾液酸类抑制剂会保护细胞免受微生物感染引起的炎症[20-21]。
laninamivir(图3)是在zanamivir基础上设计的抑制剂,是将zanamivir的2位3-羟基丙烷基团替换为1-甲氧基-2,3-二羟基丙烷基团形成的。laninamivir比zanamivir和环己烯类抑制剂oseltamivir具有更长效的抑制效果:zanamivir和oseltamivir需要1d服用2次;而laninamivir只需1d服用1次,且服用后7d内都有很强的抑制效果。laninamivir在日本已经上市,商品名为inavir[22]。

图3 laninamivir的分子结构Fig.3 Molecular structure of laninamivir
3.2 环己烯类
zanamivir的研制成功促使神经氨酸酶抑制剂的母核筛选扩展到更大的范围。以环己烯为母核的衍生物比神经氨酸的氧杂环结构更稳定,而且可以在原来氧分子的位置添加多种取代基更好地和神经氨酸酶活性中心相互作用。如将zanamivir的三羟基丙烷基团替换为脂溶性更好的烷基以利于和神经氨酸酶的Glu276发生作用。以戊烷基作为脂溶性基团的新化合物被命名为oseltamivir[23],在与 Glu276结合时位置会发生调整,并且,oseltamivir和Arg224形成1个疏水区域,正好接纳疏水性的戊烷基。oseltamivir对A型和B型流感病毒都有抑制效果,IC50值分别为1×10-9mol·L-1和3×10-9mol·L-1[24]。oseltamivir的乙酯形式在1999年被FDA批准作为口服抗病毒药物由Roche在市场上销售,商品名为tamiflu。tamiflu在体内被酯酶水解为oseltamivir的活性形式来发挥作用。研究发现神经氨酸酶的变异也对oseltamivir产生耐药性[25]。将oseltamivir 6位碳上的基团替换为含脂肪侧链后,IC50值降至10-11mol·L-1级别。这是因为,脂肪侧链的疏水性、侧链的长度、手性结构对抑制活性有很大影响[24]。将oseltamivir的醚氧基侧链替换为含氮杂环后柔性降低[26]、替换为酰胺后极性过大,均导致其不能和酶活性中心很好发生作用,抑制活性降低[27]。环己烯母核上的不同位置侧链环化形成双环化合物,也是由于刚性太强,降低了和酶活性中心的有效相互作用[28]。
3.3 环戊烷类
peramivir(图4)是以环戊烷作为母核的神经氨酸酶抑制剂。它以zanamivir的胍基和oseltamivir的戊烷基作为支链,因而可以和Glu227、Glu276相互作用。体外实验表明peramivir抑制A型和B型病毒的IC50值分别为0.1×10-9~1.4×10-9mol、0.6×10-9~11×10-9mol[29]。BioCryst制药公司对 peramivir进行Ⅲ期临床研究发现,虽然它的口服生物利用度差,但可以有效地抑制多种流感病毒。2005年美国FDA批准了peramivir注射剂的临床试验。由于peramivir具有zanamivir中4位胍基和oseltamivir中6位大位阻疏水戊烷基,所以神经氨酸酶活性位点的变异很难对peramivir产生耐药性[30]。

图4 peramivir的分子结构Fig.4 Molecular structure of peramivir
3.4 苯甲酸类
苯甲酸类神经氨酸酶抑制剂(图5)的苯环刚性太强,不能和神经氨酸酶分子的活性中心有效地契合,所以苯甲酸类化合物作为神经氨酸酶抑制剂的活性不高[31]。目前对苯甲酸类神经氨酸酶抑制剂的研究还停留在计算机模拟阶段。

图5 2种苯甲酸类神经氨酸酶抑制剂的分子结构Fig.5 Molecular structure of two analogous of benzoic acid as neuraminidase inhibitor
3.5 吡咯环类
A-525675(图6)是含吡咯环母核的神经氨酸酶抑制剂。也是参照神经氨酸和神经氨酸酶活性中心相互作用而设计的:将相当于zanamivir 5位的N-乙酰基团替换为丙烯基后,疏水性更好,有利于和Glu119、Glu227、Asp151发生相互作用[32]。利用构效定量关系模拟吡咯环化合物和神经氨酸酶的相互作用,发现氢键和分子间的吸引力(库仑引力)是主要作用力[33]。目前,A-525675的合成路线也有很多种[34-35]。

图6 A-525675的分子结构Fig.6 Molecular structure of A-525675
3.6 吲哚生物碱类
从海洋来源的链霉菌中筛选出2种吲哚生物碱类化合物,它们对神经氨酸酶的IC50值分别为67.8×10-6mol·L-1和 122.8×10-6mol·L-1[36]。siastation B是从链霉菌中分离到的1种具有神经氨酸酶抑制效果的化合物,进一步鉴定为(3S,4S,5R,6R)-6-乙酰氨基-4,5-双脱氢哌啶-3-羧酸,该化合物具有广谱的抑制效果。其合成路线有很多报道[37]。
3.7 多聚体
为了更好地使zanamivir发挥抑制作用,研究发现2个zanamivir分子连在一起的抑制效果比zanamivir单体提高100倍[8]。虽然zanamivir二聚体的生物利用度不好,但是在小鼠实验中表现出更好的抗流感功能。zanamivir二聚体在肺中比zanamivir单体具有更长的保留时间,服用7d后残留量是zanamivir单体残留量的100倍。目前这种zanamivir二聚体由日本Sankyo和澳大利亚Biota公司继续研发[38]。
将zanamivir连接到多羟基聚醚链载体上形成的多聚体比单体具有更好的抑制效果[39]。Honda等[40]将zanamivir连接到多聚谷氨酸载体上,虽然体外抑制效果变差,但小鼠实验发现多聚体有利于小肠的吸收,提高了小鼠的抗病毒能力。
3.8 融合蛋白
由于病毒的不断变异,流感病毒对oseltamivir出现耐药性。DAS181是一种具有神经氨酸酶抑制活性的融合蛋白,它含有粘性放线菌神经氨酸酶活性中心区域和人表皮锚定蛋白的结合区域[41],在大肠杆菌中成功融合表达。经过纯化后,对多种流感病毒有很强的抑制性[42]。DAS181的商品名为fludase。
3.9 多环化合物
Kirchmair等[43]通过计算机模拟筛选出5个对A、B型病毒有很好抑制效果的多环化合物,它们的IC50值为0.18×10-6~17×10-6mol·L-1,其中artocarpin(图7)可以明显抑制3株对oseltamivir敏感的病毒,也可以抑制1株对oseltamivir不敏感的病毒。

图7 artocarpin的分子结构Fig.7 Molecular structure of artocarpin
4 展望
利用神经氨酸酶的分子结构和底物的计算机契合、预测,是目前分子药物开放研究的一个良好平台,也为抑制剂的临床试验奠定了基础。目前已有2种抗流感药物投放市场,还有多种抑制剂处于临床研究阶段,神经氨酸酶抑制剂的开发范围会越来越广。
抑制剂和神经氨酸酶活性中心上Glu276的相互作用依然是研究的突破点。神经氨酸酶抑制剂可以和其它种类的抑制剂联合作用,例如离子通道抑制剂[44]、三唑核苷[45]或其它抗非病毒药物[46]。另外,筛选自然界中含有神经氨酸酶抑制剂的微生物也是近年来的研究热点,为神经氨酸酶抑制剂的开发提供了更广阔的空间。
[1]ABRASHEV I,DULGUEROVA G.Neuraminidases(sialidases)from bacterial origin[J].Experimental Pathology and Parasitology,2000,4(4):35-40.
[2]KIM S,OH D B,KANG H A,et al.Features and applications of bacterial sialidases[J].Applied Microbiology and Biotechnology,2011,91(1):1-15.
[3]MONTI E,PRETI A,VENERANDO B,et al.Recent development in mammalian sialidase molecular biology[J].Neurochemical Research,2002,27(7-8):649-663.
[4]BLIX F G,GOTTSCHARLK A,KLENK E.Proposed nomenclature in the field of neuraminic and sialic acids[J].Nature,1957,179(4569):1088.
[5]COLMAN P M,VARGHESE J N,LAVER W G.Structure of the catalytic and antigenic sites in influenza-virus neuraminidase[J].Nature,1983,303(5912):41-44.
[6]CHONG A K J,PEGG M S,TAYLOR N R,et al.Evidence for a sialosyl cation transition-state complex in the reaction of sialidase from influenza-virus[J].European Journal of Biochemistry,1992,207(1):335-343.
[7]TAYLOR N R,von ITZSTEIN M.Molecular modeling studies on ligand-banding to sialidase from influenza virus and the mechanism of catalysis[J].Journal of Medicinal Chemistry,1994,37(5):616-624.
[8]von ITZSTEIN M.The war against influenza:Discovery and development of sialidase inhibitors[J].Nature Reviews Drug Discovery,2007,6(12):967-974.
[9]XU G,RYAN C,KIEFEL M J,et al.Structural studies on the Pseudomonasaeruginosasalidase-like enzyme PA2794suggest substrate and mechanistic variations[J].Journal of Molecular Biology,2009,386(3):828-840.
[10]LEE C W,SAIF Y M.Avian influenza virus[J].Comparative Immunology Microbiology and Infectious Diseases,2009,32(4):301-310.
[11]ISLAM T,von ITZSTEIN M.Anti-influenza drug discovery:Are we ready for the next pandemic?[J].Advances in Carbohydrate Chemistry and Biochemistry,2007,61:293-352.
[12]PENG Y F,WANG X D,WEI D Z.Development of a large scale process for the conversion of polysialogangliosides to monosialotetrahexosylganglioside with a novel strain of Brevibacterium casei producing sialidase[J].Biotechnology Letters,2007,29(6):885-889.
[13]ZHANG J,CAO D,SHEN D,et al.Efficient conversion from polysialogangliosides to monosialotetrahexosylganglioside using Oerskovia xanthineolytica YZ-2[J].Bioprocess and Biosystems Engineering,2011,34(4):493-498.
[14]STREICHER H.Approaches to carbocyclic sialidase inhibitors[J].Monatshefte Fur Chemie,2002,133(10):1263-1278.
[15]NOHLE U,BEAU J M,SCHAUER R.Uptake,metabolism and excretion of orally and intravenously administered,double-labeled N-glycoloylneuraminic acid and single-labeled 2-deoxy-2,3-dehydro-N-acetylneuraminic acid in mouse and rat[J].European Journal of Biochemistry,1982,126(3):543-548.
[16]von ITZSTEIN M,WU W Y,KOK G B,et al.Rational design of potent sialidase-based inhibitors of influenza-virus replication[J].Nature,1993,363(6428):418-423.
[17]HOLZER C T,von ITZSTEIN M,JIN B,et al.Inhibition of sialidases from viral,bacterial and mammalian sources by analogues of 2-deoxy-2,3-dedihydro-N-acetylineuraminic acid modified at the C-4position[J].Glycoconjugate Journal,1993,10(1):40-44.
[18]WOODS J M,BETHELL R C,COATES J A V,et al.4-Guanidino-2,4-dideoxy-2,3-dehydro-N-acetylineuraminic acid is a highly effecitive inhibitor both of the sialidase(neuraminidase)and of growth of a wide-range of influenza A and influenza B viruses in vitro[J].Antimicrobial Agents and Chemotherapy,1993,37(7):1473-1479.
[19]GUBAREVA L V,WEBSTER R G,HAYDEN F G.Cross resistance of influenza virus mutants to NA inhibitors:Zanamivir,GS-4071,and RWJ-270201[J].Antiviral Research,2000,46(1):A54.
[20]CHEN G Y,CHEN X,KING S,et al.Amelioration of sepsis by inhibiting sialidase-mediated disruption of the CD24-SiglecG interaction[J].Nature Biotechnology,2011,29(5):428-435.
[21]PAULSON J C,KAWASAKI N.Sialidase inhibitors DAMPen sepsis[J].Nature Biotechnology,2011,29(5):406-407.
[22]IKEMATSU H,KAWAI N.Laninamivir octanoate:A new longacting neuraminidase inhibitor for the treatment of influenza[J].Expert Review of Anti-Infective Therapy,2011,9(10):851-857.
[23]KRAMER P J,ZAWADA R J X,MCDANIEL R,et al.Rational design and engineered biosynthesis of a novel 18-carbon aromatic polyketide[J].Journal of the American Chemical Society,1997,119(4):635-639.
[24]KIM C U,LEW W,WILLIAMS M A,et al.Structure-activity relationship studies of novel carbocyclic influenza neuraminidase inhibitors[J].Journal of Medicinal Chemistry,1998,41(14):2451-2460.
[25]BANTIA S,ANANTH S,HORN L,et al.Generation and characterization of a mutant of influenza A virus selected with the neuraminidase inhibitor RWJ-270201[J].Antiviral Research,2000,46(1):A60.
[26]LEW W,WU H W,CHEN X W,et al.Carbocyclic influenza neuraminidase inhibitors possessing a C3-cyclic amine side chain:Synthesis and inhibitory activity[J].Bioorganic & Medicinal Chemistry Letters,2000,10(11):1257-1260.
[27]KERRIGAN S A,SMITH P W,STOODLEY R J.Synthesis of(4R*,5R*)-4-acetylamino-5-diethylcarbamoylcyclohex-1-ene-1-carboxylic acid and (3R*,4R*)-4-acetylamino-3-diethylcarbamoylcyclohex-1-ene-1-carboxylic acid:New inhibitors of influenza virus sialidases[J].Tetrahedron Letters,2001,42(28):4709-4712.
[28]JONES P S,SMITH P W,HARDY G W,et al.Synthesis of tetrasubstituted bicyclo[3.2.1]octenes as potential inhibitors of influenza virus sialidase[J].Bioorganic & Medicinal Chemistry Letters,1999,9(4):605-610.
[29]BABU Y S,CHAND P,BANTIA S,et al.BCX-1812(RWJ-270201):Discovery of a novel,highly potent,orally active,and selective influenza neuraminidase inhibitor through structure-based drug design[J].Journal of Medicinal Chemistry,2000,43(19):3482-3486.
[30]SMITH B J,MCKIMM-BRESHKIN J L,MCDONALD M,et al.Structural studies of the resistance of influenza virus neuramindase to inhibitors[J].Journal of Medicinal Chemistry,2002,45(11):2207-2212.
[31]HULL A N,MORRILL J A.Design of benzoic acid derivatives as broad-spectrum influenza neuraminidase inhibitor using QSAR and docking[J].Abstracts of Papers of the American Chemical Society,2010,239.
[32]ZHAO C,MARING C,SUN M,et al.Design,synthesis and activity of substituted pyrrolidine influenza neuraminidase inhibitors[J].Antiviral Research,2000,46(1):A53.
[33]SUN J,MEI H.Docking and 3D-QSAR investigations of pyrrolidine derivatives as potent neuraminidase inhibitors[J].Chemical Biology & Drug Design,2012,79(5):863-868.
[34]DEGOEY D A,CHEN H J,FLOSI W J,et al.Enantioselective synthesis of antiinfluenza compound A-315675[J].Journal of Organic Chemistry,2002,67(16):5445-5453.
[35]HANESSIAN S,BAYRAKDARIAN M,LUO X H.Total synthesis of A-315675:A potent inhibitor of influenza neuraminidase[J].Journal of the American Chemical Society,2002,124(17):4716-4721.
[36]江宏磊,王传喜,江宁宇,等.海洋来源链霉菌FIM090041产生的神经氨酸酶抑制剂[J].中国抗生素杂志,2012,37(4):265-268.
[37]KNAPP S,ZHAO D L.Synthesis of the sialidase inhibitor siastatin B[J].Organic Letters,2000,2(25):4037-4040.
[38]MACDONALD S J F,CAMERON R,DEMAINE D A,et al.Dimeric zanamivir conjugates with various linking groups are potent,long-lasting inhibitors of influenza neuraminidase including H5N1avian influenza[J].Journal of Medicinal Chemistry,2005,48(8):2964-2971.
[39]WATSON K G,CAMERON R,FENTON R J,et al.Highly potent and long-acting trimeric and tetrameric inhibitors of influenza virus neuraminidase[J].Bioorganic & Medicinal Chemistry Letters,2004,14(6):1589-1592.
[40]HONDA T,YOSHIDA S,ARAI M,et al.Synthesis and anti-influenza evaluation of polyvalent sialidase inhibitors bearing 4-guanidino-Neu5Ac2en derivatives[J].Bioorganic & Medicinal Chemistry Letters,2002,12(15):1929-1932.
[41]MALAKHOV M P,ASCHENBRENNER L M,SMEE D F,et al.Sialidase fusion protein as a novel broad-spectrum inhibitor of influenza virus infection[J].Antimicrobial Agents and Chemotherapy,2006,50(4):1470-1479.
[42]TRIANA-BALTZER G B,GUBAREVA L V,KLIMOV A I,et al.Inhibition of neuraminidase inhibitor-resistant influenza virus by DAS181,a novel sialidase fusion protein[J].PLoS One,2009,4(11):e7838.
[43]KIRCHMAIR J,ROLLINGER J M,LIEDL K R,et al.Novel neuraminidase inhibitors:Identification,biological evaluation and investigations of the binding mode[J].Future Medicinal Chemistry,2011,3(4):437-450.
[44]松本庆藏,王镇山,万献尧.神经氨酸酶抑制剂[J].日本医学介绍,2004,25(7):310-313.
[45]SMEE D F,BAILEY K W,MORRISON A C,et al.Combination treatment of influenza A virus infections in cell culture and in mice with the cyclopentane neuraminidase inhibitor RWJ-270201 and ribavirin[J].Chemotherapy,2002,48(2):88-93.
[46]CARTER M J.A rationale for using steroids in the treatment of severe cases of H5N1avian influenza[J].Journal of Medical Microbiology,2007,56(7):875-883.
