Grp78基因过表达慢病毒载体构建和包装及鉴定*
2014-08-14李亚文徐世元张庆国赖露颖苏娇玲杨耐梅李元涛
李亚文,徐世元,张庆国,李 乐,赖露颖,郑 艇,苏娇玲,杨耐梅,李元涛
(1.南方医科大学珠江医院麻醉科,广州 510282;2.南方医科大学附属深圳妇幼保健院麻醉科,广东深圳 518028)
长期以来,过表达microRNA质粒可直接用于细胞系转染表达,是研究基因功能的重要方法。但用真核细胞构建的表达载体在细胞转染阶段存在转染率不高且周期长等弊端,限制其广泛应用;而慢病毒载体是以人类免疫缺陷型病毒(HIV)为基础发展起来的基因治疗载体,它既能感染分裂细胞又能感染非分裂细胞,安全性高并可以在体内较长期地表达。本实验所用慢病毒载体感染目的细胞后不再感染其他细胞,也不会利用宿主细胞产生新的病毒颗粒。
本研究通过构建Grp78基因过表达慢病毒表达载体,为下一步感染SH-SY5Y细胞株,进而研究Grp78在布比卡因神经毒性所致细胞凋亡和细胞损伤中的作用奠定基础。
1 材料与方法
1.1 材料 (1)主要试剂:琼脂糖、PCR试剂盒、Taq polymerase,dNTP、限制性内切酶、质粒抽提试剂盒。台盼蓝、胰酶、DMSO、DMEM、Lipofectamine 2000、细胞株:293T;菌株:大肠埃希菌菌株DH5α;病毒载体:GV 载体、pHelper 1.0载体、pHelper 2.0载体。(2)主要仪器:PCR仪、稳压DNA电泳仪、凝胶成像仪、培养箱、高速离心机、荧光显微镜、CO2培养箱。
1.2 方法
1.2.1 PCR扩增目的基因 (1)载体信息:GV261;元件顺序:Ubi-MCS-IRES-Cherry;克隆位点:AgeI/NheI。(2)目的基因片段Grp78引物,上游:5′-CGG GTA CCG GTC GCC ACC ATG AAG CTC TCC CTG GTG G-3′;下 游:5′-AGT CGC TAG CCT ACA ACT CAT CTT TTT CTG CTG TAT C-3′。
1.2.2 制备感受态细胞及转化 CaCl2法制备感受态细胞进行转化实验步骤如下:(1)各取每种感受态细胞悬液200μL转移至无菌微量离心管中,每管加入连接液10μL,轻轻旋转以混匀,然后置冰中放置30min。制备感受态细胞,使其具有摄取外源DNA的能力。(2)42℃热休克90s。快速将管转移到冰浴中冷却细胞1~2min。每管加入800μL LB培养基。水浴加温至37℃,然后放置摇床温育45min以复苏细菌。(4)将150μL转化感受态细胞转移到AMP(100μg/mL)抗性的LB琼脂培养基上。把平板置于室温直至液体被吸收。然后倒置平皿,置于37℃培养箱中培养16h。(5)克隆进行后续PCR鉴定。
1.2.3 重组质粒构建 PCR产物连接入线性化表达载体反应体系见表1,反应条件 :25℃30min;42℃15min。
1.2.4 Lentivirus病毒包装 消化293T细胞,调整其密度为每20mL有1.2×107个细胞,接种细胞于培养皿中,放置37℃,5%CO2培养箱中培养24h,待细胞密度达70%~80%时可用于细胞转染。细胞状态对于病毒包装至关重要,需要保证良好的细胞状态和较少的传代次数。转染前2h将含胎牛血清培养基更换为无血清培养基。向一离心管中加入制备的各DNA溶液(pGC-LV 载 体20μg、pHelper 1.0 载体15μg、pHelper 2.0载体10μg)与相应体积培养基混合均匀,调整总体积为2.5mL,室温下温育5min。将Lipofectamine 2000试剂轻柔摇匀,取100μL Lipofectamine 2000试剂在另一管中与2.4mL Opti-MEM培养基混合,室温下温育5min。把稀释后的DNA与稀释后的Lipofectamine 2000进行混合,轻颠混匀,避免振荡,且须5min内混合。混合后室温下温育20min,然后将混合液转移至293T细胞培养液中,混合均匀,放置37℃、5%CO2培养箱中培养。培养8h后倒去培养基,每瓶细胞加入20mL磷酸盐缓冲液(PBS),以洗涤残余混合液,移去混合液。每瓶细胞中加入含10% 胎牛血清培养基25mL,放置37℃,5%CO2培养箱内继续培养2d。

表1 PCR反应体系
1.2.5 Lentivirus滴度测定
1.2.5.1 样品制备 293T细胞传代,24孔中每个孔加入1×105个细胞,体积为500μL;次日准备10个无菌Ep管,每管中加90μL培养基;取待测病毒原液10μL加入到第一个管中,混合均匀,取混合均匀的第一管液10μL加入到第二个管中继续相同的操作直到最后一管;选取所需细胞孔,吸去90μL培养基。加稀释好的病毒溶液,放置于37℃,5%CO2培养箱培养;1d后,加入新鲜培养基500μL。小心操作;4d后抽提RNA。
1.2.5.2 总RNA抽提 去细胞上清液,每孔加入1mL Trizol,吹打,室温静置5min,转移至另一新1.5mL Ep管中。每管加200μL氯仿,用力震荡15s,室温下静置15min。4℃下12000r/min离心15min。从每管中吸取上清液至另一新1.5 mL Ep管中。加入等体积-20℃预冷的异丙醇,混匀后-20℃沉淀10min。4℃下12000r/min离心10min,移去上清液。加入1mL 4℃预冷的75%乙醇,洗涤沉淀及离心管壁。4℃下10000r/min离心5min,移弃上清液。4℃下10000 r/min再次离心5min,吸去残液,室温下干燥,不需完全干燥。加20μL无RNA酶(RNase)水至完全溶解,紫外分析测定所抽提RNA浓度。
1.2.5.3 RNA逆转录获cDNA 将1μL Oligo dT(0.5μg/μL)和2.0μg总RNA加入PCR小管,补焦碳酸二乙酯(DEPC)水至9μL。混合均匀、离心,70℃温浴10min。紧接着插入到0℃冰水浴中。按下表比例,根据反应管数算出所需试剂量。将M-MLV酶等在冰上混匀得到逆转录反应液。在每个反应管中加11μL逆转录反应液,混合均匀后离心。其中,11μL逆转录反应液含5×逆转录缓冲液4μL、10mmol/L dNTPs 2μL、RNA 抑制剂 0.5μL、M-MLV-RTase 1μL、DEPC水3.5μL。在42℃进行1h完成逆转录反应,后用70℃处理10min使逆转录酶失活。逆转录反应产物cDNA可用于PCR,也可-80℃长期保存。
1.2.5.4 实时定量PCR检测 实时定量PCR在Takara的TP800PCR仪上完成。配置反应体系:每管加入SYBR premix ex Taq 10μL、上游引物(5μmol/L)1.0μL、下游引物(5μmol/L)1.0μL、cDNA 1.0μL、ddH2O 7.5μL。设定程序为两步法实时定量PCR。预变性95℃5s;变性95℃5s,退火60℃30s,延伸60℃30s,40个循环。每次在延伸阶段读取吸光值,用于制作熔解曲线。PCR结束后,在95℃变性1 min。然后冷却至55℃,使DNA双链充分聚合。从55℃开始到95℃,每一步增加0.5℃,保持30s,同时读取吸光值。两个循环后将调定点升高0.5℃。
2 结 果
2.1 重组质粒构建结果 阳性转化子PCR产物大小:510 bp,见图1。
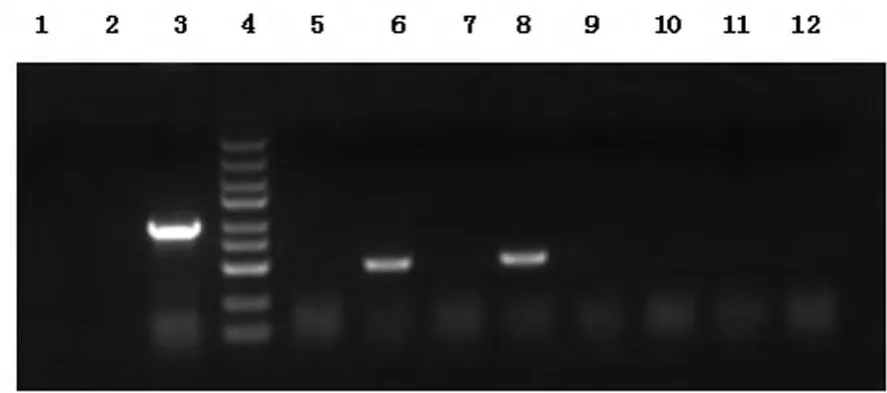
图1 PCR鉴定转化结果电泳图
2.2 病毒的收获及浓缩 收集转染后2d的293T细胞上清液。4℃、4000×g离心10min,以除去细胞碎片。用0.45 μm过滤器过滤上清液到40mL超速离心管中。把病毒粗提液样品加入过滤杯中。将过滤杯插到滤过液收集管中,再4000×g离心至所需病毒浓缩体积,时间15min。离心结束后取出离心装置,将过滤杯和滤过液收集杯分开。将过滤杯倒扣在样品收集杯上,离心力不超过1000×g,时间2min。过高转速会使样品损失。把过滤杯从样品收集杯上移开。样品收集杯中即为病毒浓缩液。将病毒浓缩液移出,进行分装;取其中一支进行滴度测定,其余置-80℃长期保存。
2.3 实时定量PCR法测定绿色荧光蛋白标记的慢病毒滴度
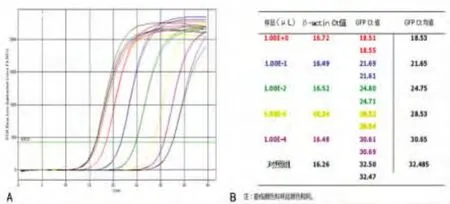
图2 实时定量PCR曲线图
2.3.1 引物信息 绿色荧光蛋白(GFP)引物:被扩增的片段位于211~496bp之间,其上游引物序列为5′-TGC TTC AGC CGC TAC CC-3′,熔点Tm为57.4℃,GC百分含量为64.7%。下游引物序列为5′-AGT TCA CCT TGA TGC CGT TC-3′,熔点Tm为57.3℃,GC百分含量为50.0%。β-actin被扩增的片段位置位于932~1233bp之间,其上游引物序列为5′-GTG GAC ATC CGC AAA GAC-3′,熔点 Tm 为52.6℃,GC百分含量为55.6%。下游引物序列为5′-AAA GGG TGT AAC GCA ACT A-3′,熔点Tm为51.0℃,GC百分含量为42.1%。
2.3.2 实时定量PCR结果 在该次病毒滴度检测中,1.00E-03μL组样品与对照组样品的Ct值差异大于3以上,故认定病毒颗粒存在于1.00E-03μL组样品中。见图2。
2.4 β-actin、GFP熔解曲线 见图3。

图3 β-actin、GFP熔解曲线
3 讨 论
通过慢病毒载体的构建、包装,得到稳定表达的质粒;该质粒能有效应用于研究Grp78基因过表达来减少细胞内质网应激反应[1]。该质粒较采用真核细胞转染得到的质粒感染细胞的成功率高[2-3]。观察 GFP表达情况[4-5],在加入 1E-5μL病毒原液的孔中观察到3个细胞存活,说明该孔中至少有3病毒颗粒感染细胞,且认为该病毒的滴度等于带有荧光的细胞数除以病毒原液量。在加入1E-6μL病毒原液的孔中观察到2个带有荧光的细胞,说明该孔中至少有2个病毒颗粒感染细胞,且认为该病毒的滴度等于带有荧光的细胞数除以病毒原液量,即2/(1E-6)=2E+6,单位为 TU/μL,也就等于2E+9 TU/mL。
加入不同病毒量的细胞样品,通过提取总RNA后反转录为cDNA,然后进行定量PCR检测,通过对照组与实验组的Ct值差异来判断滴度值。通常情况下认为Ct值差异2以上存在显著差异[6]。反转录反应所获得20μL cDNA中只取1μL用于实时定量PCR检测,该结果仅表示1/20样品的情况,所以在滴度计算时乘以系数20。
退火温度偏低,可引起非特异性扩增。适当提高退火温度可降低非特异性扩增。故通过测量升高温度后荧光的变化可以帮助降低非特异产物的影响。非特异性条带的出现,原因诸多,引物与靶序列不完全互补、或引物聚合形成二聚体、退火温度过低、PCR循环次数过多等。针对解决措施有降低引物量、减少循环次数、适当提高退火温度或采用二温度点法(93℃变性,65℃左右退火与延伸)。熔解曲线中,由于SYBR GreenⅠ与所有双链DNA相结合,引物二聚体、单链二级结构以及错误的扩增产物引起的假阳性会影响定量精确性。故由熔解曲线来分析产物的均一性更有助于准确分析SYBR Green实时定量 PCR 结果[7-13]。
设立适当的阳性对照和阴性对照,阳性对照以能出现扩增条带的最低量标准病原体核酸为宜,注意交叉污染可能。每次反应都应有一管不加模板的试剂对照及相应不含有被扩增核酸的样品作阴性对照。PCR结束后,根据发生过程中荧光值变化绘出每个样品的熔解曲线。熔解曲线是扩增反应的质控途径,图中没有出现杂峰,也未出现主峰的异常增宽,表明实验中未出现污染、引物二聚体和非特异性扩增。不同的扩增产物因为其长度和GC含量不同而在不一样的温度下解链,当产物解链时,SYBR GreenⅠ的荧光值将降低并被仪器所监测。绘制熔解曲线时,需实时定量PCR仪连续监测每个样品在从双链完全配对到完全解链的升温过程中荧光值的变化,由此绘制出荧光强度随温度变化的负一次倒数图。荧光强度变化的拐点(熔点,Tm)即为熔解峰值。Grp78过表达病毒载体的构建和包装成功,为今后研究Grp78基因在布比卡因所致神经细胞损伤中的作用奠定基础,进而为布比卡因神经毒性的防治打开一扇科学之门。
[1]李亚文,徐世元,张庆国,等.高糖环境诱导SH-SY5Y细胞ROS爆发-内质网应激增强布比卡因神经毒性[D].广州:南方医科大学,2013.
[2]Huang P,He Z,Ji S,et al.Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors[J].Nature,2011,475(7356):386-389.
[3]Anokye-Danso F,Trivedi CM,Juhr D,et al.Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency[J].Cell Stem Cell,2011,8(4):376-388.
[4]Wang L,Zhou GB,Liu P,et al.Dissection of mechanisms of Chinese medicinal formula Realgar-Indigo naturalis as an effective treatment for promyelocytic leukemia[J].Proc Natl Acad Sci U S A,2008,105(12):4826-4831.
[5]Zheng J,Shen WH,Lu TJ,et al.Clathrin-dependent endocytosis is required for TrkB-dependent Akt-mediated neuronal protection and dendritic growth[J].J Biol Chem,2008,283(19):13280-13288.
[6]Zhang H,Feng W,Liao W,et al.The gp130/STAT3signaling pathway mediates beta-adrenergic receptor-induced atrial natriuretic factor expression in cardiomyocytes[J].FEBS J,2008,275(14):3590-3597.
[7]Sun Y,Liu M,Yang B,et al.Role of siRNA silencing of MMP-2gene on invasion and growth of laryngeal squamous cell carcinoma[J].Eur Arch Otorhinolaryngol,2008,265(11):1385-1391.
[8]Zhang L,Liu HJ,Li TJ,et al.Lentiviral vector-mediated siRNA knockdown of SR-PSOX inhibits foam cell formation in vitro[J].Acta Pharmacol Sin,2008,29(7):847-852.
[9]Federico M.Lentivirus gene engineering protocols[J].Methods Mol Biol,2003,30(4):458-460.
[10]Tiscornia G,Singer O,Verma IM.Production and purification of lentiviral vectors[J].Nat Protoc,2006,1(1):241-245.
[11]Sena-Esteves M,Tebbets JC,Steffens S,et al.Optimized large-scale production of high titer lentivirus vector pseudotypes[J].J Virol Methods,2004,122(2):131-139.
[12]Reiser J.Production and concentration of pseudotyped HIV-1-based gene transfer vectors[J].Gene Ther,2000,7(11):910-913.
