p110δ突变失活上调血管MMPs表达并引发血管壁结构损伤*
2014-08-13邢丽英郑凌云吴玉娥曾翠玲桂亚丽方海燕王丽京李卫东
邢丽英, 郑凌云, 吴玉娥, 曾翠玲, 桂亚丽, 方海燕, 吴 腾, 王丽京, 李卫东
(广东药学院 1血管生物学研究所,2基础学院,4健康学院, 3广东省实验动物监测所,广东 广州 510006)
磷酯酰肌醇3-激酶(phosphatidylinositol 3-kinase, PI3K)家族,参与如细胞生长、细胞增殖、细胞分化、细胞运动性、细胞存活和胞内运输等一系列细胞功能活动,发挥重要的生物学作用。PI3Ks能特异性地催化磷酯酰肌醇3位羟基磷酸化, 产生具有第二信使作用的肌醇脂物质的激酶,也被称为磷脂酰肌醇3-激酶或磷酸肌醇3-激酶[1]。p110δ属于该家族成员中的一个催化亚基,是一种调节机体免疫功能的催化酶, 在T细胞、B细胞、肥大细胞和嗜中性粒细胞中发挥着重要的作用,近年来p110δ作为预防或治疗炎症和自身免疫及移植排斥反应的研究靶点被广泛地进行研究[2]。
近年来的研究显示,PI3K家族中p110γ的缺失会影响PI3K激酶的信号通路和Akt的活化,从而进一步抑制氧化型低密度脂蛋白和炎症趋化因子的形成,从而减轻动脉粥样硬化斑块[3]。这说明,PI3K信号通路在血管炎症中起到一个很重要的作用。有文献报道,p110δ在血管组织中也有一定的表达,并且参与调节血管平滑肌细胞的L-Ca2+电流、内皮细胞与白细胞的黏附以及高血压进程中血管的收缩反应[4]。
动脉管壁较厚,通过弹性板分为3层同心膜。最里层的内膜由内皮细胞组成,起到减少血流阻力的作用;中膜较厚,主要由一层环状的平滑肌细胞(smooth muscle cells, SMCs)构成,可维持血管系统张力,且含弹性纤维和胶原纤维,大动脉以弹力纤维为主,中、小动脉以平滑肌为主;外膜由疏松结缔组织构成,含胶原纤维和弹性纤维,可起到防止血管过度扩张的作用[5]。动脉壁的结构与其功能密切相关。血管的结构改变可能会阻碍动脉的缓冲作用,从而引发一系列的心血管疾病,如动脉粥样硬化、高血压等,同时吸烟、动脉粥样硬化和高血压等因素引起的血管损伤又是诱发动脉瘤的关键[6]。
这些结果提示p110δ在血管重构过程中可能也有重要意义。有鉴于此,本研究采用p110δ基因突变失活(p110δ-mutant)小鼠,试图观察p110δ突变后对主动脉血管的作用,进一步探索如何通过PI3K信号通路影响血管损伤以及作用机制的研究。
材 料 和 方 法
1 实验动物
C57BL/6 (C57) 小鼠20只,雄性,8周龄,购于广东省实验动物中心,许可证号为SYXK(粤)2013-0002。p110δ-mutant小鼠20只,雄性,8周龄,购自Jackson实验室。
2 试剂和仪器
乙醇、甲醇、异丙醇和苏木素-伊红染色套装(康龙生物);琼脂糖(Biowest);PCR引物(Invitrogen);DAB显色试剂盒(Thermo);PCR Mix(Roche);RNA提取试剂盒(TaKaRa);弹性纤维染色相关试剂和胶原纤维染色相关试剂(本实验室制备);胶原蛋白(collagen)Ⅰ、collagen IV、α-平滑肌肌动蛋白(α-smooth muscle actin, α-SMA)、CD31(Abcam);内参照IgG2b由中科院上海细胞生物研究所提供;HRP标记Ⅱ抗购自EMD Calbiochem。
PCR仪(ABI);光学显微镜(Olympus);低温离心机 (Thermo);超速离心机 (Beckman);制冰机(北京长流科学仪器有限公司);凝胶成像仪(GE); 电泳仪(Bio-Rad);照相机(Canon);旋转混合仪(宁波新芝生物科技有限公司);微波炉 (Galanz ); 体视显微镜(重庆奥特光学仪器有限公司);超纯水系统(Millipore)。
3 方法
3.1提取血管组织 8周龄的雄性p110δ-mutant(p110δ-/-)纯合子小鼠20只为实验组,同时取同周龄的野生型(wild type,WT)小鼠20只为对照组。取出胸主动脉血管,体视镜下分离,固定包埋切片。
3.2免疫组化和免疫荧光检测 从-80 ℃ 取出冰冻切片,室温放置30 min,PBS洗涤。0.01 mol/L柠檬酸盐缓冲液修复10~15 min。冷却至室温,PBS洗涤。10% BSA封闭, 37 ℃ 放置1 h。倾去10% BSA,加Ⅰ抗,4 ℃过夜。PBS洗涤。擦干玻片,在组织上加Ⅱ抗,放置1 h。组织切片晾干,滴加中性树胶,封片。
3.3免疫印迹实验检测蛋白水平 将样品与5×loading buffer混匀后100 ℃ 煮10 min,待用。配置分离胶和浓缩胶,常温凝胶1 h。SDS-PAGE分离蛋白,转膜250 mA、4 °C 90 min。将PVDF膜置于含5%脱脂奶粉的TBST中,室温封闭1 h。TBST洗3次,每次5 min。加Ⅰ抗: 4 ℃孵育过夜。TBST室温下脱色,加Ⅱ抗,室温下孵育1 h后,用TBST在室温下脱色摇床上洗3次,每次10 min。配置发光液,等体积均匀混合溶液A和溶液B于洗膜盒,约2~3 mL。将膜倒扣于发光液上,反应5 min后将膜包于保鲜膜中,注意避免产生皱折和气泡,放入X光片夹中。在暗室中,将X光片放在膜上,压片时间一般视不同抗体而定,显影,定影。 凝胶图像分析:将胶片进行扫描或拍照,Quantity-One凝胶蛋白分析软件对条带进行分析。
3.4Real-time PCR检测动脉血管中MMPs的mRNA表达 动脉血管总RNA提取采用Trizol法,以1 μg总RNA为模版逆转录合成cDNA第一链,以此cDNA为模版进行PCR反应。PCR反应在LightCycler PCR 2.0仪器(Roche)中进行。基因相对表达量用2-ΔΔCT法分析。
4 统计学处理
数据用均数±标准差(mean±SD)表示,采用SPSS 13.0软件对数据统计进行单因素方差分析,以P<0.05为差异有统计学意义。
结 果
1 小鼠基因型鉴定结果
3号鼠和6号鼠的PCR产物电泳结果可见602 bp和425 bp 条带,为p110δ杂合子(基因型为p110δ+/-),5号鼠和7、8、9号鼠的PCR 产物电泳结果仅见602 bp条带,为p110δ纯合子(基因型为p110δ-/-),1、2号鼠和4号鼠的PCR产物电泳结果可见425 bp条带,提示其为野生型(基因型为p110δ+/+),见图1。

Figure 1. PCR results of p110δ gene for identification of p110δ- mutant mice. Lanes 1, 2, 4 were p110δ+/+; Lanes 3, 6 were p110δ+/-; Lanes 5, 7, 8, 9 were p110δ-/-; M was the marker.
2 p110δ突变失活后血管的形态学染色
光镜下观察结合对各组动脉血管直径测量统计图显示p110δ突变失活后小鼠的主动脉血管壁变薄,管腔变大。从HE染色、EVG染色和Gomori醛品红染色中看出C57小鼠血管结构较完整,未发生明显变化,p110δ突变后,与正常对照组相比,血管壁的三层结构部分破坏,弹性纤维出现较多的褶皱,见图2。
3 p110δ突变失活后血管内皮细胞的变化
如图3A所示,p110δ突变后,血管的内皮细胞层不完整,这可能是由于内皮细胞的凋亡引起的;进一步做了CD31和TUNEL的免疫荧光共定位,发现p110δ突变后血管内皮细胞的凋亡明显增多,说明血管内皮细胞的结构和功能均发生了损伤。如图3B所示,根据血管中凋亡细胞情况所做的统计学分析,p110δ突变后血管内皮细胞的凋亡明显高于C57组(P﹤0.05)。

Figure 2. The pathological characteristics of the thoracic aorta in p110δ-mutant mice. A, B: vascular wall become thinner and vessel diameter enlarged after inactivating mutation of p110δ; C: smooth muscle cell vacuolar degeneration, morphological disorders, elastic fiber local fracture. Mean±SD. n=6.*P<0.05 vs C57.
4 p110δ突变失活后血管平滑肌细胞、胶原的表达
如图4所示,与C57小鼠相比,p110δ突变小鼠血管壁的α-SMA表达降低,细胞外基质中胶原蛋白Ⅰ和Ⅳ减少,说明血管壁结构受到了严重破坏。
5 p110δ突变失活后血管基质金属蛋白酶(matrix metalloproteinase,MMP)的变化
p110δ突变后主要引起了MMP-3和MMP-9的mRNA表达上调,其中MMP-3上调尤为明显;金属蛋白酶组织抑制因子1(tissue inhibitor of metalloprotei-nase 1,TIMP-1)和TIMP-2水平上调。p110δ突变失活后很可能由于影响了MMPs的水平而造成了血管壁结构损伤,见图5。
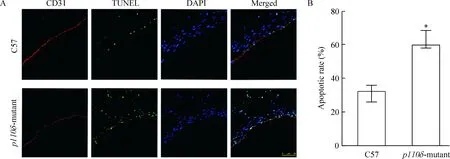
Figure 3. The vascular endothelial cell apoptosis and degeneration in p110δ-mutant.A: vascular endothelial cells (CD31) positioning with TUNEL; B: statistics of apoptotic rate in the blood vessels. Mean±SD.n=6.* P<0.05 vs C57.

Figure 4. The levels of collagen contents in vascular smooth muscle cells were reduced after inactivating mutation of p110δ. A: the IHC images of vascular paraffin section to observe the expression of α-SMA, collagenⅠ and collagen Ⅳ. Scale bar=20 μm. B: the protein levels extracted from fresh aorta determined by Western blotting. Mean±SD.n=6.*P<0.05 vs C57.
讨 论
PI3K是一种具有脂激酶活性的酶类家族,共分为3 大类。其中I 类的PI3K是由催化亚基(p110)和调节亚基(p85/p87/p101)构成的异源二聚体,催化亚基p110 又有4种亚型p110α、p110β、p110γ和p110δ。p110 亚基的共同特点是磷酸化细胞膜的磷脂酰肌醇,产生第二信使PtdIns(3,4,5)P3,进而募集并激活蛋白激酶B和其它诸如GSK3、Raf 等的信号分子,从而广泛参与细胞的生长、分化和免疫调节等作用。不同的p110 亚基组织分布差异很大,其中p110δ主要在白细胞中表达,参与T、B 淋巴细胞的分化、成熟以及中性粒细胞的化学趋化过程,因此p110δ一直被认为是白细胞中调控免疫反应的重要分子之一[7]。近年来的研究还显示,p110δ在血管组织中也有一定的表达,并且参与调节血管平滑肌细胞的L-Ca2+电流、内皮细胞与白细胞的黏附以及高血压进程中血管的收缩反应[8]。这些结果提示可能p110δ在血管损伤过程中也有重要意义,但具体的作用和机制不明。
血管重构的病理机制与血管壁慢性炎症、MMPs活化以及氧化应激等密切相关。MMPs是一组能特异降解细胞外基质的酶家族,主要降解血管壁的弹力纤维和胶原蛋白,破坏血管结构的稳定性,在组织重构中起重要作用[9]。 MMPs/TIMPs 的动态平衡维持着血管的正常形态和功能,TIMP 作为MMP 的抑制因子, 其水平随MMP 的变化而变化[10]。MMPs的表达调控主要体现在转录水平[11],因此我们做了real-time-PCR实验检测MMPs和TIMPs的mRNA水平。实验结果MMP-3、MMP-9的mRNA表达上调,其中MMP-3上调尤为明显;TIMP-1、 TIMP-2水平上调。MMP-3能够降解胶原Ⅳ和弹力纤维,也可以激活其它蛋白酶如MMP-9,增强MMP-9的降解Ⅳ型胶原的能力,介导平滑肌迁移和血管内膜新生[12],可见MMP-3在血管重构中有着关键作用。MMP-3和MMP-9水平升高,很可能是诱发血管壁弹力纤维降解和胶原蛋白含量降低的重要原因。MMP-2降解基底膜的主要成分胶原Ⅳ,在血管重构中起到的作用已得到证实, MMP-14能活化MMP-2,并与TIMP-2相互作用[13],3者如何共同作用对血管结构产生影响还需要进一步研究。而TIMP 通过与MMPs 活性形式结合的方式而抑制其作用, MMPs 和TIMPs 之间的平衡是MMP 活性一个关键控制点[14]。所有的MMPs一旦被活化都将受到TIMP的抑制作用,但是MMP-2和MMP-9前体和TIMPs结合反而有利于前体的活化[15],能协同参与降解血管壁细胞外基质。

Figure 5. The expression of Matrix metalloproteinase (MMPs) was up-regulated after inactivating mutation of p110δ.Mean±SD.n = 6.*P<0.05,**P<0.01 vs C57.
研究表明,血管平滑肌细胞和炎症细胞是血管重构中MMPs的主要来源。已有文献表明MMP-2和MMP-9可通过潜在转化生长因子β(transforming growth factor β, TGF-β)复合物的蛋白水解激活TGF-β[16],MMPs水平升高可能通过升高TGF-β水平诱发血管壁炎症,影响血管重构。转录因子激活蛋白-1(activator protein-1, AP-1)参与调节炎症因子在血管重构中的作用,在细胞增殖和炎性反应中发挥重要作用。研究发现AP-1可在动脉粥样硬化形成过程中对各种促动脉粥样硬化炎症因子发挥调节作用,从而可以对动脉粥样硬化的形成及进展过程进行调控。磷脂酰肌醇3-激酶信号通路与其主要的下游效应分子蛋白激酶B参与细胞分化、增殖、凋亡以及迁徙等,可通过调节AP-1等转录因子结合到特定的启动子或促进子上进而提高炎症介质基因的转录活性,最终导致细胞产生大量细胞因子,因而PI3K/Akt信号转导通路在机体炎症反应调控过程中起着极其重要作用。然而目前关于PI3K/Akt/AP-1通路在血管平滑肌细胞信号的表达,以及PI3K/Akt通路对血管炎症作用的研究较少。MMP-2、TIMP-1 基因上有AP-1 的结合位点,TGF-β1、TNF-α基因中也含有AP-1 的结合位点[17-18], AP-1 失活很可能影响TGF-β1、TNF-α的合成及其功能, 间接调节MMP-2 的分泌与活化。在本实验中,p110δ失活很可能通过调控PI3K/Akt/AP-1信号通路影响了MMPs的表达,进一步影响了主动脉的结构和功能,此猜测还需进一步实验验证。
综上所述,p110δ突变失活后小鼠动脉血管结构异常,可观察到血管中的内皮细胞凋亡,平滑肌细胞结构损伤,细胞空泡变性,弹力纤维局部断裂,免疫组化染色发现α-SMA在血管平滑肌细胞中的表达降低,胶原含量减少,进一步实验发现p110δ突变失活后上调了MMPs的表达,我们推断p110δ突变失活后很可能通过上调MMPs的蛋白表达,降解了血管中的细胞外基质成分,从而损伤血管结构,但是如何上调的,还在探索当中。以上p110δ突变失活通过上调MMPs引发血管壁损伤的研究,为我们对人类心血管疾病的研究提供了一条新线索,通过进一步研究p110δ对人类心血管疾病的影响,可以检测其是否能成为诊断人类心血管疾病的指标之一。
[参 考 文 献]
[1] Fougerat A, Gayral S, Malet N, et al. Phosphoinositide 3-kinases and their role in inflammation: potential clinical targets in atherosclerosis?[J]. Clin Sci (Lond), 2009, 116(11): 791-804.
[2] Harris SJ, Foster JG, Ward SG. PI3K isoforms as drug targets in inflammatory diseases: lessons from pharmacological and genetic strategies[J]. Curr Opin Invest Drugs, 2009, 10(11):1151-1162.
[3] Wymann MP, Solinas G. Inhibition of phosphoinositide 3-kinase γ attenuates inflammation, obesity, and cardiovascular risk factors[J]. Ann N Y Acad Sci, 2013, 1280: 44-47.
[4] Geng L, Tan J, Himmelfarb E, et al. A specific antagonist of the p110delta catalytic component of phosphatidylinositol 3’-kinase, IC486068, enhances radiation-induced tumor vascular destruction[J]. Cancer Res, 2004, 64(14):4893-4899.
[5] Swift MR, Weinstein BM. Arterial-venous specification during development[J]. Circ Res, 2009, 104(5):576-588.
[6] Krecki R, Kasprzak JD. Advanced peripheral arterial disease in a 59-year-old man with suspected acute coronary syndrome and normal coronary angiogram[J]. Postepy Kardiol Interwencyjnej, 2013, 9(3):307-309.
[7] Deane JA, Fruman DA. Phosphoinositide 3-kinase: diverse roles in immune cell activation[J]. Annu Rev Immunol, 2004, 22:563-598.
[8] Kobayashi T, Hayashi Y, Taguchi K, et al. ANG II enhances contractile responses via PI3-kinase p110 delta pathway in aortas from diabetic rats with systemic hyperinsulinemia[J]. Am J Physiol Heart Circ Physiol, 2006, 291(2):H846-H853.
[9] Van Lint P, Libert C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation[J]. J Leukoc Biol, 2007, 82(6):1375-1381.
[10]Hire JM, Evanson JL, Johnson PC, et al. Variance of matrix metalloproteinase (MMP) and tissue inhibitor of metalloproteinase (TIMP) concentrations in activated, concentrated platelets from healthy male donors[J]. J Orthop Surg Res, 2014, 9:29.
[11]Yan C, Boyd DD. Regulation of matrix metalloproteinase gene expression[J]. J Cell Physiol, 2007, 211:19-26.
[12]Johnson JL, Dwivedi A, Somerville M, et al. Matrix metalloproteinase (MMP)-3 activates MMP-9 mediated vascular smooth muscle cell migration and neointima formation in mice[J]. Arterioscler Thromb Vasc Biol, 2011, 31(9):e35-e44.
[13]Tetu B, Brisson J, Wang CS, et al. The influence of MMP-14, TIMP-2 and MMP-2 expression on breast cancer prognosis[J]. Breast Cancer Res, 2006, 8(3):R28.
[14]Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis[J]. Genes Dev, 2000, 14(2):163-176.
[15]Jinga DC, Blidaru A, Condrea I, et al. MMP-9 and MMP-2 gelatinases and TIMP-1 and TIMP-2 inhibitors in breast cancer: correlations with prognostic factors[J]. J Cell Mol Med, 2006, 10(2):499-510.
[16]Brew K, Dinakarpandian D, Nagase H. Tissue inhibitors of metalloproteinases: evolution, structure and function[J]. Biochim Biophys Acta, 2000, 1477(1-2): 267-283.
[17]Dhandapani KM, Hadman M, De Sevilla L, et al. Astrocytes protect ion of neurons: role of transforming growth factor-beta signaling via a c-Jun-AP-1 protective pathway[J]. J Biol Chem, 2003, 278(44):43329-43339.
[18]Cho MK, Jang YP, Kim YC, et al. Arctigenin, a phenylpropanoid dibenzylbutyrolactone lignan, inhibits MAP kinases and AP-1 activation via potent MKK inhibit ion: the role in TNF-α inhibition[J]. Int Immunopharmacol, 2004, 4(10-11):1419-1429.
