生物类似物政策监管指南原则与要求研究综述
——基于欧盟版生物类似物指南
2014-08-10陈名邵蓉
陈 名 邵 蓉
1.中国药科大学国际医药商学院 江苏南京 211198
2.中国药科大学国家药物政策与医药产业经济研究中心 江苏南京 211198
生物类似物政策监管指南原则与要求研究综述
——基于欧盟版生物类似物指南
陈 名1,2*邵 蓉1,2
1.中国药科大学国际医药商学院 江苏南京 211198
2.中国药科大学国家药物政策与医药产业经济研究中心 江苏南京 211198
本文通过文献综述,在明确界定生物类似物概念的基础上,阐述了生物药的特点及其研发生产过程。基于欧盟版生物类似物指南,介绍了其政策框架,并系统梳理了生物类似物政策监管指南原则与要求,包括生物相似性、安全性和免疫原性、适应症外推法、标签与命名、数据保护以及药物互换性与药物警戒等内容,旨在为促进我国生物类似物与生物制药产业的进一步发展提供政策参考与建议。
生物类似物; 欧盟; 欧洲药品管理局; 监管指南; 技术要求
近年来,一些生物药品的专利陆续到期,为全球制药企业带来了巨大的研发机遇和市场空间,未来生物类似物将在市场上扮演更为重要的角色。而与传统化学药物相比,生物类似物因其生产工艺的严格、复杂、精细性,难以保障化学制药中仿制药与原研药物间的生物等效性,仅能与原研生物药之间存在高度相似性。传统意义上将生物类似物等价为生物仿制药,实际上缺乏一定科学依据。
因此,欧盟药品管理局于2004年起草生物类似物指南,这是世界上首个建立生物类似物监管指南的机构。该指南于2005年正式生效,首次对生物类似物进行明确的概念界定,提出生物类似物不是仿制药。经过近十年的发展,美国、加拿大、澳大利亚、日本等国家已纷纷起草并制定了生物类似物指南的框架文件,规范和促进生物类似物的研发,满足医药行业日益高涨的研发诉求。2013年,我国国家食品药品监督管理局药品审评部门也开始进行生物类似物指导原则的调研和起草等工作,预计于2014年底出台首版生物类似物指南。本文基于欧盟版生物类似物指南,运用文献研究方法梳理生物类似物政策监管指南研究内容,为促进我国生物类似物与生物制药产业的进一步发展提供政策参考与建议。
1 生物类似物的定义与分类
1.1 定义
在欧盟、WHO和美国已出台的生物类似物指南中,欧盟(指南CHMP/437/04)将生物类似物术语界定为“与已通过审批的参比药物相似的生物药品(Similar Biological Medicinal Product or Biosimilar)”[1],并明确指出生物类似物不是仿制药。WHO指南参照欧盟的定义,采用治疗性生物制品(Similar Biotherapeutic Products,SBPs)来界定生物类似物。[2]美国定义生物类似物为“与FDA批准的参比药物相比具有高度相似性或可替代其的生物药品”。加拿大则采用“随后进入的生物制品(Subsequent Entry Biologics,SEBs)”[3]来直观表明生物类似物的出现在时间上落后于原研药物。澳大利亚则采用与欧盟一致的术语。[4- 5]日本采用“后续生物医药产品(Follow-on Biological Medicinal Product, FOBMP)”界定生物类似物[6](表1)。

表1 部分国家与国际组织关于生物类似物的术语界定
目前大量文献研究中提及生物类似物采用的是欧盟和美国的定义。由于仿制药的标准(生物等效性和相同的成分)无法适用于生物类似物研究,因此从科学角度针对类似物采用生物仿制药(Biogeneric)这一术语并不精确,易造成理解误区。表2将生物类似物与Me-too生物药、Bio-better进行概念区分。随着越来越多生物药品的专利到期,应当明确生物类似物的概念界定并规范与使用术语,以应用于生物药品的研究。我国未来生物医药产业的发展,需注重生物类似物的研发。

表2 生物类似物、Me-too生物药和Bio-better的定义[7]
1.2 分类
按照欧美已获批生物类似物的活性物质分类,可将生物类似物分为红细胞生成素(Epoetin,EPO)、重组白细胞生成素(Filgrastim,G-CSF)、人体生长激素(Somatropin,rh-GH)、干扰素(IFN)和单克隆抗体(Monoclonal Antibodies,mAbs)等类别。其中,红细胞生成素又分为多种形式,如EPO-alfa、-beta,-delta,-omega和-zeta等。欧盟作为世界上最早发布生物类似物指南的地区,也是世界上生物类似物获批数量最多的地区[8],获批生物类似物以EPO和Filgrastim/G-CSF两类产品为主,各类别产品审批(通过/撤回/拒绝)的数量情况见表3。

表3 2005—2014年欧盟各类生物类似物审批数量情况
2 生物类似物的特点及其研发生产过程
2.1 特点
2.1.1 分子特性导致不能采用化学仿制药的生物等效性验证方法
与化学药相比,生物药的分子往往由复杂的蛋白质结构组成,分子量相对庞大(表4)、大多具备三维空间结构与较低的稳定性,导致其分子特性极易受到光、热、温度等环境因素影响。因而在研发过程中,生物药的活性成分难以与原研药完全一致,不能按照化学仿制药研发中与原研药具有相同化学成分的标准进行研发,即生物类似物的研发不能够运用生物等效性来验证[9],欧盟生物类似物指南中也明确指出了这一点。
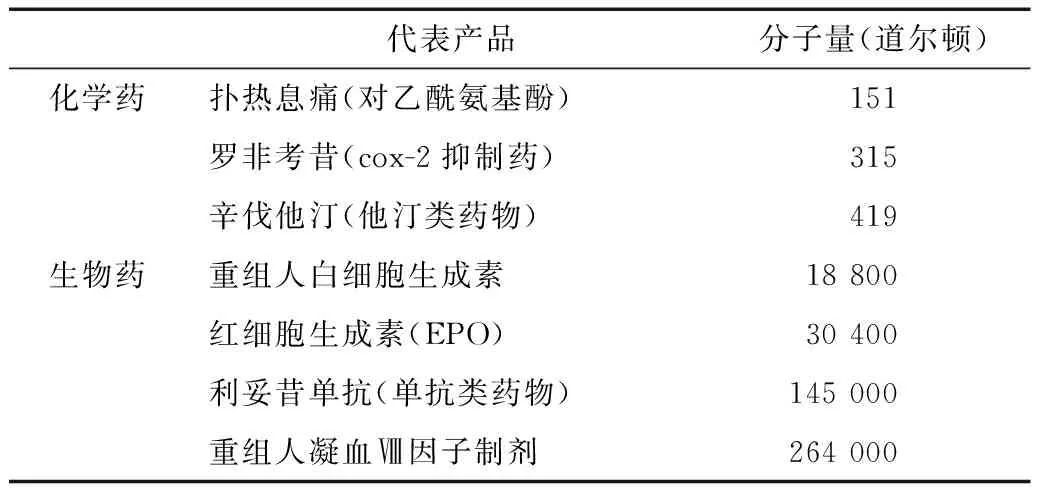
表4 一些典型化学药与生物药的分子量对比[10]
2.1.2 采用极为严格精细的药品生产工艺保证最终产品质量属性
在生产方面,与化学药相比,生物药的生产更为精细与复杂,最终产品的属性很大程度取决于生产工艺。以市面上占比较多的蛋白类生物药为例(以下均以此为例),蛋白质生产中独特的细胞表达系统涉及宿主细胞的选择,不同的宿主细胞以及后期生产中蛋白质的纯化与验证、储存与包装等条件的不同都有可能造成其三维空间结构、羧基数量和糖基化等方面的改变,从而影响产品的质量属性。
2.2 研发生产过程
生物类似物分子的蛋白质属性决定它独特精细的生产过程,不同蛋白质来源、分离及纯化过程都会导致产品的异质性。蛋白质的生产过程主要分为两方面:(1)DNA基因克隆和蛋白质表达;(2)蛋白质的产生、纯化及验证。具体而言,将一段DNA序列中所含目的基因克隆至表达载体,再将载体转染至宿主细胞,通过不同的细胞表达进行转录与翻译形成蛋白质表达;细胞在不同的培养条件和增殖方式下,在生物反应器进行扩增,经过滤、离心、色谱等方法纯化蛋白,最终形成具备特征属性的纯化的原料产品。[11]
由于知识产权因素,生物类似物的生产商不能获得参比药物生产过程的具体信息,增加了蛋白质产品被精确复制的难度。现有分析技术的受限,也导致蛋白质生物学和临床特性不能被完全预测。面对这些客观受限因素,如何促进生物类似物的研发、保障生物类似物的安全有效性,亟需出台规范合理的生物类似物研发标准与指导原则。
3 生物类似物的监管原则与要求
为了科学有效地促进生物类似物的研发以及保障患者用药安全,欧盟药品管理局(EMA)在2005年版生物类似物指南基础上不断更新、修订与完善,相继发布一系列相关总则以进一步完善生物类似物的审批与监管(表5),旨在满足日益增长的生物类似物研发诉求,尤其是单克隆抗体的研发,也针对临床研究中的一致性评价、试验设计、生物标记物和外推法等内容进行修订。本研究基于生物类似物指南,对监管的原则和要求进行详细介绍。

表5 2005年以来欧盟生物类似物指南及系列总则
3.1 生物相似性
生物等效性是针对同一种药物的不同制剂,在相同实验条件下给予相同剂量,判断其吸收速度和程度有无显著差异的过程。在化学药物研发中,可用来比较已上市药物的新剂型与原剂型是否生物等效(Equivalence),或判断仿制药与创新药是否具有同等(Identical)的安全有效性。但由于生物类似物难以与原研药有效成分一致,类似物生产商也无法获取原研药生产过程信息,增加了产品一致性的难度。因此,传统化学仿制药研发途径不适于生物类似物,而应证明生物类似物与原研药间的生物相似性(Similarity)。
3.1.1 合理选择参比药物
依照欧盟指令2001/83/EC第八款规定,生物类似物的参比药物品应是已获得上市许可的药物。[12]对于生物类似物生产商,应当优先选择具备完整的质量、安全性与有效性评价数据的药品作为参比药物,最好该药已上市并被使用了一定时间。[12-14]在药物剂型与给药途径方面,生物类似物与参比药物需保持一致。在研发初期,需紧密结合企业研发策略与预期产品适应症,慎重、择优选择参比药物,以规避与降低不慎选择参比药物对后期比较试验的研发风险与资本消耗。对于批准参比药物的药监部门,在生物治疗药物的评价方面应当具备较为丰富的实践经验与完善的监管措施。
3.1.2 科学验证生物类似物与参比药物之间的生物相似性
(1)质量相似性研究。质量相似性研究决定着生物类似物后期非临床与临床研究需提供数据的多少程度。影响生物类似物与参比药物质量相似性的因素主要有生产过程和药品特性(如物化性质、生物活性、免疫特性等)。生产过程中,生产商应严格遵循药品生产质量管理规范(GMP)设置质量保障和控制系统,最大程度地优化生产过程以减少在验证质量相似性过程中可能引起的误差,同时减少可预测范围内影响生物类似物安全性与有效性的不良因素。在药品特性的比较性研究中,应当采用合适的生化和生物分析方法探寻生物类似物和参比药物的特性。在物化特性研究方面,可采用色谱分析方法决定蛋白质的原始结构和二级、三级乃至四级结构。在生物活性研究方面,应结合临床研究,反映出蛋白质的反应机理,检测出具备活性的物质,发现生物类似物和参比药物之间的重要功能性区别。考虑到药物的免疫特性(如抗体与抗原),也应注重比较药物的特异性、亲和性和功能活性等。在其它方面,应采取先进的技术探测与药物生产及自身产品相关的杂质,若在生物类似物和参比药物间观测到显著差异,应分析其是否会对安全性、有效性以及免疫原性造成影响。
(2)药物的非临床研究。欧盟生物类似物指南(EMEA/CHMP/BMWP/42832/2005)规定,生物类似物的非临床研究应参照《生物技术来源药物的非临床安全性评价指南注意事项(CHMP/ICH/302/95)》进行。生物类似物的非临床研究由体外(In Vitro)和体内(In Vivo)研究两部分构成。体外研究应运用受体结合研究和细胞水平检测研究,以阐释生物类似物与参比药物间的比较性。体内研究应设计合理的动物实验与模型,利用已知的药物相关信息进行生物类似物与参比药物的比较性研究,使研究结果能有效服务于临床研究。其中,非临床毒性研究应保证足够的时间以便对生物类似物与参比药物间在毒性与免疫反应方面的不同进行检测。通常,安全药理学、生殖毒理学、致突变性和致癌性研究等常规的毒理学研究不适用于生物类似物非临床研究。
(3)药物的临床研究。生物类似物的临床研究需进行药代动力学(PK)研究、药剂学研究(PD)和临床试验研究。药代动力学研究通常在参比药物的给药途径及其治疗剂量范围之内实施,在同质人群中采取单剂量交叉试验以发现生物类似物与参比药物间的不同。通常采用最敏感的剂量来获得最佳的测量效果。在伦理范畴内,应在健康志愿者人群中进行药代动力学研究,若所研药物具备已知不良反应,并在药理作用和风险方面被认为不适于健康志愿者人群,则研究应在患者人群中进行。而药剂学研究常与药代动力学研究联合进行,并提供使用剂量和疗效的相关信息。对于比较性药剂学研究,通常选择剂量效应曲线上的最陡峭部分对应的剂量[15]应用于合适人群。在实际监管方面,针对不同类型的生物产品,如胰岛素、生长激素等,EMA采用不同的非临床与临床指南严格评价与管理生物类似物。[16]
3.2 安全性和免疫原性
生物药品的免疫原性受细胞株选择、杂质、给药途径、剂量范围、蛋白质稳定剂以及患者、疾病与治疗等因素影响,药物生物活性增强或降低,进而引起免疫反应或并发症等不良反应[17],具有不可预测性[5,18]。表6列出了近年来全球范围内因生物药品研发生产过程中的一些变化所引发的治疗问题与不良反应症状,这表明了药物免疫原性研究与安全性之间存在关联,也对生物药品生产商提出了更为严谨与慎重的研发要求。

表6 药物研发生产中某些变化导致生物药品免疫原性与安全性隐患的情况[4,19-20]
3.3 适应症外推法
如果生物类似物与参比药物在某一适应症上具有高度相似性且相关适应症的反应机制相同,那么可将生物类似物的适应症范围外推扩张至这些相关适应症。这一过程需得到临床经验和相关研究数据的支持,同时也存在一定安全隐患。对于生物药品生产商,在适应症外推过程中,应明确采用哪些数据。[22]
3.4 标签与命名
明确的药品标签与命名是直观区分生物类似物与原研药的首要途径。[23]国际药品通用名(简称“通用名”)通常被用于化学合成的分子药物,但由于生物类似物无法与原研药具有相同的活性成分,该命名并不能反映出生物类似物的药品特性,因此,应采用合理的命名方式从根本上减少对生物类似物及生物药品的认识误区。在生物类似物的命名及标签信息方面,生产商应确保所提供的信息完整、无误,应包含产品信息(如试验数据、安全性评价等)[11],还应标明与原研药的区别以及二者是否可替换使用,并提供市场上其它同质产品信息。目前,欧盟获批的生物类似物已具备其独特的商品名信息。
3.5 数据保护
药品数据保护是药品知识产权保护的一种,旨在药品审批上市后的一定周期内不受理仿制药申请,给予原研药在该周期内的市场独占权利,激励药物创新。[24]然而,数据保护的意义也同样适用于生物药。目前,欧盟生物类似物指南对于数据保护的规定为“8+2+1”数据独占期,具体而言,其中2年为原研药的市场独占期,1年为对于已有产品新适应症的保护期。[25]WHO规定依照各具体地区的相关指南进行。美国FDA将给予原研生物药12年的数据独占期以极大地激励生物药的创新[8,26],若药品用于儿科适应症还将增加6个月的期限。
3.6 药物互换性与药物警戒
药物互换性指在临床治疗时具有相似疗效的生物类似物与原研药间的相互替换。在药品使用环节,当考虑成本较低的生物类似物作为对应原研药的临床治疗新选项时,不仅应关注二者间的安全性、有效性,还应当重视二者间的互换性。
欧盟现有生物类似物指南未规定药物互换性方面内容,药学层面上不允许生物类似物与原研药相互替换。欧盟各个国家具体的药物互换性规定也需由本国药品管理当局制定,其中,英国、法国、德国、西班牙等国家不允许生物类似物与原研药互换使用。[5,11,17]WHO对于药物互换性尚未进行具体规定。美国FDA在《生物制剂价格竞争与创新法》(BPCI Act)中要求生物类似物与原研药具有高度相似性并在任一患者中能够产生相同治疗效果,当生物药品的使用超过一次,采用生物类似物治疗所产生的安全性与有效性风险应当不超过使用相关原研药的风险。[27]
目前,有学者对此提出质疑,认为“任一”患者与“相同”治疗效果的前提难以在现实中达成,那么针对生物类似物与原研药间的互换性,可能通过生物统计学模型的评价方法更为科学合理,依此将互换性分为互换(Switching)与交替(Alternating)使用,旨在通过构建生物相似性指数、互换指数与交替指数等模型予以评价。[28]关于生物类似物的互换性,世界上各国家与地区药品管理当局仍在探寻一种科学合理的指导原则。
药物警戒系统通过制定药品评价预案以监测与预防药品上市后可能的不良反应以及安全性问题[17],保障药品的可追溯性,EMA也提倡风险管理计划。通常上市前的生物药品临床数据难以充分验证药品一些潜在的安全特性,为保障患者用药安全,上市后的药品监管(包括对免疫原性的研究)与药物警戒系统设置极为必需,这也要求各国药品管理当局协同合作,合理沟通,提供及时、准确、有效的监管数据,共同完善生物药品临床使用数据库。
4 小结
本文通过系统梳理世界上生物类似物政策监管指南原则与要求内容,建议我国在生物类似物指南的制定过程中,首先应当明确生物类似物的概念,以规避生物仿制药的认识误区。遵循保障药品安全、有效、质量可控的基本原则,积极探寻生物类似物监管指南框架的可行性,在参照欧盟、WHO或美国等国家和地区的指南时,应当立足我国国情,与现行药事法规衔接。此外,在满足我国制药企业研发诉求的同时,也应当从促进高质量生物类似物研发和生物医药健康持续发展的长远角度,力求保持与国际上接轨,为生物制药的全球化提供政策支持。
[1]European Medicines Agency (EMA).Concept Paper on the Revision of the Guideline on Similar Biological Medicinal Product[Z].London: EMA, 2011.
[2]World Health Organization (WHO).Guidelines on Evaluation of Similar Biotherapeutic Products (SBPs)[R].Geneva: WHO, 2009.
[3]Health Canada.Guidance for Sponsors: Information and Submission Requirements for Subsequent Entry Biologics (SEBs)[R].Ottawa: Health Canada, 2010.
[4]Kaur Chugh P, Roy V.Biosimilars: Current Scientific and Regulatory Considerations[J].Current clinical pharmacology, 2014, 9(1): 53- 63.
[5]Jha D, Mishra R K, Pandey R.Biosimilars: Current regulatory perspective and challenges[J].Journal of pharmacy & bioallied sciences, 2013, 5(1): 80.
[6]Ministry of Health, Labour and Welfare (MHLW).Guidelines for the Quality, Safety and Efficacy Assurance of Follow- On Biologics[R].Tokyo: MHLW, 2009.
[7]Weise M, Bielsky M C, De Smet K, et al.Biosimilars [mdash]why terminology matters[J].Nature biotechnology, 2011, 29(8): 690-693.
[8]Carey K.Biosimilars encircle Rituxan, US debates innovator exclusivity[J].Nature biotechnology, 2011, 29(3): 177- 178.
[9]Schellekens H, Ryff J C.‘Biogenerics’: the off- patent biotech products[J].Trends in pharmacological sciences, 2002, 23(3): 119- 121.
[10]Roger S D.Biosimilars: How similar or dissimilar are they?(Review Article)[J].Nephrology, 2006, 11(4): 341- 346.
[11]Mellstedt H, Niederwieser D, Ludwig H.The challenge of biosimilars[J].Annals of oncology, 2008, 19(3): 411- 419.
[12]European Medicines Agency (EMA).Guideline on Similar Biological Medicinal Products(CHMP/437/04).[EB/OL].[2014-01-22].http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003517.pdf
[13]Gecse K B, Khanna R, van den Brink G R, et al.Biosimilars in IBD: hope or expectation?[J].Gut, 2013, 62(6): 803- 807.
[14]Minghetti P, Rocco P, Cilurzo F, et al.The regulatory framework of biosimilars in the European Union[J].Drug discovery today, 2012, 17(1): 63- 70.
[15]杨焕.关于生物仿制药临床评价的探讨[J].中国临床药理学与治疗学, 2009, 14(1): 5- 9.
[16]Tsiftsoglou A S, Ruiz S, Schneider C K.Development and regulation of biosimilars: Current status and future challenges[J].BioDrugs, 2013, 27(3): 203- 211.
[17]Moorkoth S, Vasantharaju S G, Laggyshetty S.Indigence for Robust Pharmacovigilance in Biosimilars: A review[J].Elixir Pharmacy, 2013, 56: 13460- 13464.
[18]Ebbers H C, Crow S A, Vulto A G, et al.Interchangeability, immunogenicity and biosimilars[J].Nature biotechnology, 2012, 30(12): 1186- 1190.
[19]Revers L, Furczon E.An introduction to biologics and biosimilars.Part II: Subsequent entry biologics: Biosame or biodifferent?[J].Canadian Pharmacists Journal/Revue des Pharmaciens du Canada, 2010, 143(4): 184- 191.
[20]Kuhlmann M, Covic A.The protein science of biosimilars[J].Nephrology Dialysis Transplantation, 2006, 21(suppl 5): v4- v8.
[21]Schellekens H, Jiskoot W.Eprex- associated pure red cell aplasia and leachates[J].Nature biotechnology, 2006, 24(6): 613- 614.
[22]Suh S K, Park Y.Regulatory guideline for biosimilar products in Korea[J].Biologicals, 2011, 39(5): 336- 338.
[23]Nowicki M.Basic facts about biosimilars[J].Kidney and Blood Pressure Research, 2007, 30(5): 267- 272.
[24]丁锦希, 王颖玮, 贺晓雪, 等.药品试验数据保护制度中的新化学实体界定问题研究——基于美国 Actavis 公司诉 FDAVyvanse 案的实证分析[J].中国新药与临床杂志, 2012, 31(11): 652- 657.
[25]Simoens S, Verbeken G, Huys I.Market access of biosimilars: not only a cost issue[J].Oncologie, 2011, 13(5): 218- 221.
[26]Osborne R.Brand biologics grab 12 years’ exclusivity, for now[J].Nature biotechnology, 2009, 27(8): 677- 678.
[27]Endrenyi L, Chang C, Chow S C, et al.On the interchangeability of biologic drug products[J].Statistics in medicine, 2013, 32(3): 434- 441.
[28]Chow S C, Yang L Y, Starr A, et al.Statistical methods for assessing interchangeability of biosimilars[J].Statistics in medicine, 2013, 32(3): 442- 448.
(编辑 赵晓娟)
Reviewofbiosimilarregulatoryguidelinesandscientificprinciples:ExperiencesfromEuropeanMedicinesAgency(EMA)guidelines
CHENMing1,2,SHAORong1,2
1.DepartmentofSocialandAdministrativePharmacy,SchoolofInternationalPharmaceuticalBusiness,ChinaPharmaceuticalUniversity,NanjingJiangsu211198,China
2.ResearchCenterofNationalDrugPolicy&Ecosystem,ChinaPharmaceuticalUniversity,NanjingJiangsu211198,China
In this study, a literature review was adopted to specify terminology of biosimilars and demonstrate the basic characteristic of biologics and relevant research and development (R&D) procedures.The regulatory framework of the European Medicines Agency (EMA) guidelines on biosimilars was introduced.Explicitly, regulatory guidelines and scientific principles, regarding biosimilarity, safety and immunogenicity, extrapolation, labels and names, data protection, were systematically introduced, as well as interchangeability and pharmacovigilance, respectively.The purpose of the study is to provide regulatory references for Chinese legislators and recommendations on the R&D of biosimilars in the biopharmaceutical industry.
Biosimilars; European Union; European Medicines Agency; Regulatory guidelines; Scientific principles
陈名,女(1989年—),硕士,主要研究方向为医药政策与法规。E-mail:mindychen007@163.com
邵蓉。E-mail:shaorong118@163.com
R197
A
10.3969/j.issn.1674-2982.2014.10.005
2014-08-10
2014-09-15
