冠醚化学交换分离锂同位素的影响因素分析及进展
2014-08-08严峰袁威津李建新何本桥程煜崔振宇
严峰,袁威津,李建新,何本桥,程煜,崔振宇
(1天津工业大学中空纤维膜材料与膜过程省部共建国家重点实验室培育基地,天津300387;2天津工业大学 环境与化学工程学院,天津300387;3天津工业大学 材料科学与工程学院,天津300387)
人口的增长以及经济的高速发展日益加剧着人们对能源的需求。由于当前85%的能源来源于不可再生的化石能源如石油、天然气和煤炭等,潜在的能源危机以及环境污染的问题促使人们寻找新型清洁能源[1]。因此,开发少污染,无长寿命放射性废物,资源无限丰富的核聚变能越来越受到世界各国高度关注,我国也针对核聚变能作出了开发第四代核电技术包括钍基熔盐堆核能系统的重大战略规划。锂同位素在钍基熔盐堆核能系统发展过程中具有极其重要的作用,是必需的核燃料和冷却剂。一方面,在核反应堆运行过程中,核聚变反应是利用氘(D)和氚(T)聚合成氦(4He)时放出中子(其轰击钍232可以增殖为铀233),并释放巨大能量[2][方程式(1)]。其中,氘在海水中储量极为丰富,1L海水里提取出的氘(约33mg)通过聚变反应时可释放出相当于燃烧300L汽油的能量。但是,自然界中几乎不存在氚,而轻的锂同位素6Li被中子轰击后可以生成氚和氦,同样释放大量能量[式(2)]。所以,6Li是必不可少的核聚变堆原料。另外,重的同位素7Li由于其极小的吸收截面(0.037b)被用来作为核聚变反应堆的堆心冷却剂和导热的载热剂,7Li还可以作为钍堆熔盐介质。
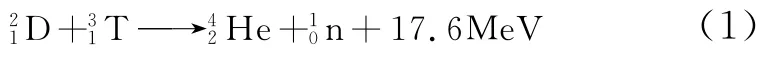
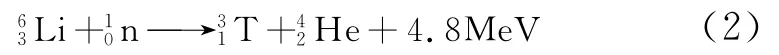
由此可见,锂同位素核聚变所需的重要原料。锂同位素分离是核能开发必需解决的关键技术,关系到国家能源安全和战略的实施。
锂是世界上最轻的金属,蕴藏量丰富,我国是第三大锂资源生产国。据估算,1kg锂含有的能量大约相当于4000t标准煤,至少可以发电10MWh。天然锂中含6Li 7.5%,7Li 92.5%,6Li与7Li的性质差异称为锂同位素效应[3]。Thewlis[4]证明6Li的离子半径大于7Li,其差值Δr(6Li-7Li)为4×10-2pm。基于锂同位素效应,锂同位素分离的方法大致可分为化学法和物理法。物理法包括熔盐电解法、电磁法、激光分离等。化学法包括锂汞齐法、溶剂萃取、离子交换色层分离等。其中,锂汞齐法[5]是唯一在工业上已获得应用的方法,其优点在于锂同位素分离系数大(基本在1.05左右);同位素交换速率极快(在剧烈逆流条件下半交换期只有几秒)。然而,锂汞齐法因使用大量剧毒汞而导致严重的环境和安全问题,欧美等国用此法分离锂同位素的工厂已部分关闭,正积极处理由此产生的大规模汞污染。因此,各国研究者们正在积极寻找其他更为安全高效的锂同位素分离方法。
自从Pedersen[6]发现冠醚类化合物,并发现冠醚化合物能根据其环的大小选择性地与金属离子尤其是碱金属离子络合以来,基于冠醚的化学交换法分离锂同位素的研究逐渐成为人们研究的热点。冠醚类化合物是一类人工合成的受体,典型冠醚是由有机连接体(如—CH2CH2—基团)连接的醚氧原子所组成的简单排列,冠醚环中具有不均匀的电荷分布。图1所示为计算机模拟的12-冠-4的电荷分布模型[7],红色区域代表负电荷区域,绿色代表中性区域,蓝色代表正电荷区域。从图1可以看出,冠醚分子外层显示荷正电性,冠醚分子内侧由于氧原子的未共用电子呈现负电性,因此凭借离子-偶极相互作用,可与金属离子产生主-客体结合,在其大环空穴内与金属离子形成稳定的络合物(也有环外夹心式的结合模式)[8]。
Jepson等[9]首先研究了锂盐-穴醚[2,2,1]交换体系,测得单级分离系数α=1.041±0.006,该体系的同位素效应可与汞齐体系相比较。之后日本大阪大学的Nishizawa等[10]采用液-液萃取获得12-冠-4对锂同位素分离因子为1.057,韩国忠北大学的Kim等[11]以N3O3杂氮冠醚螯合树脂为固定相,采用淋洗色谱法获得锂同位素分离系数达1.068,伊朗核科学与技术研究所的Davoudi等[12]采用分散液-液微萃取方法获得苯并15-冠-5对锂同位素分离因子高达1.070。由此可见,冠醚类化合物分离锂同位素有很好的分离效应,单从分离系数看,它是目前最有前途的锂同位素分离方法。
冠醚应用于锂同位素分离主要可划分为两种方法:液-液萃取和固-液萃取。液-液萃体系分离锂同位素是指将冠醚和锂盐分别溶解于互不相溶的有机相和水相,利用锂同位素6Li和7Li在两相间的不同分配,经过反复多次萃取获得锂同位素富集的方法。液-固体系一般指将冠醚接枝到高分子链段制备冠醚聚合物材料,再通过柱式色谱的方法分离锂同位素。无论是冠醚小分子还是冠醚聚合物,由于产生离子络合的有效结构都是冠醚受体,所以它们具有相同的锂同位素分离机理。本文将结合文献阐释不同因素条件(冠醚分子环结构、配位阴离子效应、萃取温度、溶剂效应)对冠醚化学交换体系(液-液萃取法和色层法)锂同位素分离效应的影响规律。
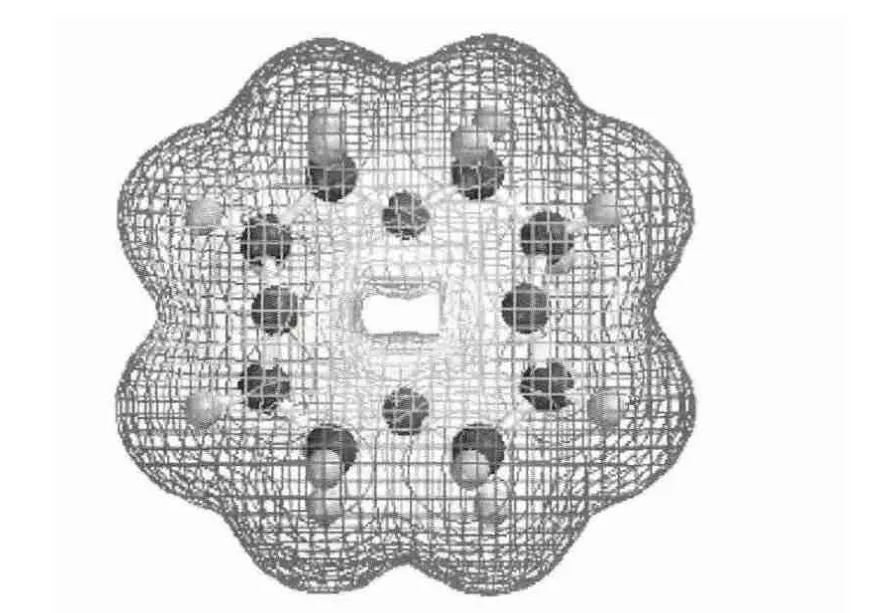
图1 计算机模拟的12冠4的模型[7]
1 冠醚受体分离锂同位素原理
1.1 冠醚络合锂离子的热力学过程
在冠醚络合锂离子的过程中,体系自由能将发生变化。冠醚络合锂离子的净自由能变ΔG°由体系焓变ΔH°和熵变ΔS°共同组成,具体来说,净自由能变指冠醚-锂络合的焓与游离溶剂分子增加所获得的熵的和,减去冠醚与锂离子去溶剂化以及冠醚构象重排引起焓的损耗[13]。在络合过程的不同阶段,许多因素影响着冠醚络合锂离子过程的自由能变ΔG°(图2)。由图2可以看出,冠醚和锂离子在互相络合前首先经历自身去溶剂化的过程,即过程(1)和(2)。去溶剂化需要吸热,对ΔG°不利,溶剂分子的剔除增加体系熵,这对ΔG°有利。过程(1)和(2)的主要影响因素包括了溶剂种类。冠醚在与锂离子络合的过程中会出现构象的变化[过程(3)],这过程出现焓损耗,对ΔG°不利。这就需要引入冠醚结构的设计,以便降低构象重排产生额外能量损失。过程(4)中,冠醚与锂离子形成络合物,该过程释放能量,体系变得更加稳定,是络合过程的主要推动力,对ΔG°高度有利。过程(4)受到诸多因素的影响,如冠醚给体原子种类、冠醚侧链以及配位阴离子种类等。最后,溶剂分子对冠醚-锂络合物依然存在溶剂化效应,并且影响着络合物的稳定性,见过程(5)。在各种因素互相影响下最终形成了冠醚受体在某一体系的锂同位素分离效应。
自由能变的绝对值越大意味着络合力越大。冠醚的锂同位素分离效应本质上是冠醚对6Li、7Li络合自由能变的差异引起的。由此可见,影响冠醚锂同位素分离效应的因素不仅包括冠醚自身结构如冠醚环大小、给体原子种类、侧链种类等,化学交换过程的工艺条件如锂盐配位阴离子种类、溶剂种类、化学交换温度等也将产生显著影响。
1.2 冠醚分离锂同位素各评价参数定义
由以上分析可以指出,冠醚锂同位素分离效应源于两个交换平衡反应包括式(3)和式(4)的自由能变ΔG°的差异[14]。

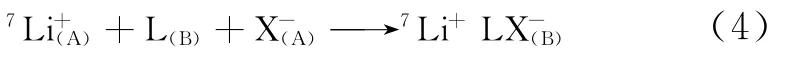
式中,L代表冠醚,X-代表阴离子,下角标A代表溶剂相,下角标B代表冠醚存在相。
理论上,式(3)和式(4)的自由能变以及各自的熵变和焓变都可以独立获得,两式对应的自由能变ΔG°差异越大则意味着分离因子的提升。但是以上数据需要通过冠醚与纯6Li和7Li的热力学实验获得,具有较高实验要求,所以在锂同位素化学交换体系,自由能变、熵变和焓变主要通过式(5)获得。
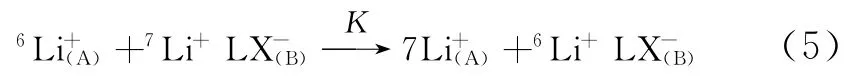
式(5)的平衡常数K可以定义如式(6)。
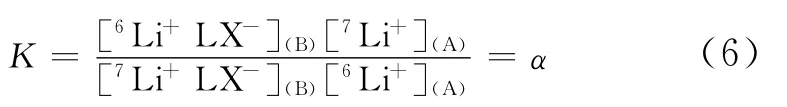
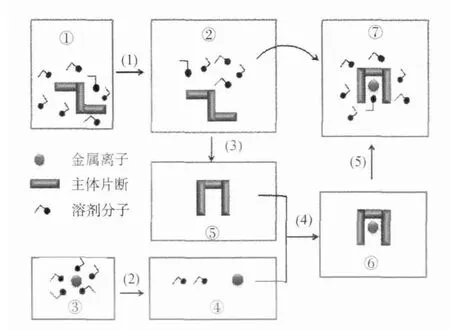
图2 主客体络合过程
单级分离因子α拥有与平衡常数K一样的定义,所以分离因子即为K值。
为了更全面了解冠醚的锂同位素分离效应,在实际研究过程需要引入其他参数用以表征。比如在液-液萃取中,冠醚的锂同位素分离效应的表征参数有萃取率(Extractability)、有机相络合度(P)及锂同位素分离因子α1等,液-固体系中的冠醚聚合物的锂同位素分离效应的表征参数有锂吸附量A以及分离因子α2来表征,对应的定义如式(7)~式(10)。
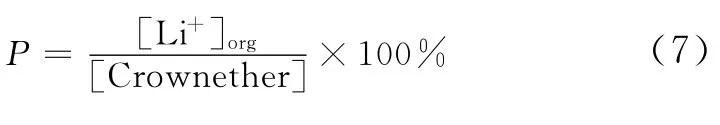
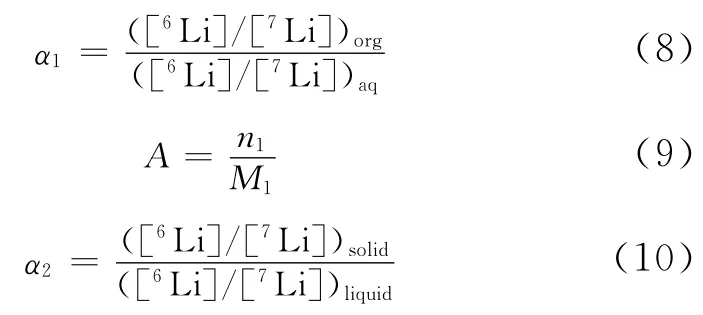
式中,n1为络合锂的物质的量,mmol;M1为冠醚固载化合物的质量,g。
以下将结合文献数据详细阐述冠醚化学交换体系中不同的条件因素对锂同位素分离效应的影响规律。
2 冠醚分子结构对锂同位素分离效应的影响
冠醚通过化学交换分离锂同位素的过程中,分离效应的影响因素主要来源两个方面,一方面来自冠醚受体本身的分子结构,另一方面来自化学交换过程的工艺条件。首先对冠醚分子结构的方面进行锂同位素分离效应的阐述。
2.1 分子尺寸效应
不同的冠醚分子结构(图3)拥有不同的孔腔大小,孔腔大小影响着冠醚对锂离子的络合作用力以及图3锂同位素分离因子。表1为碱金属离子直径以及部分冠醚孔径。
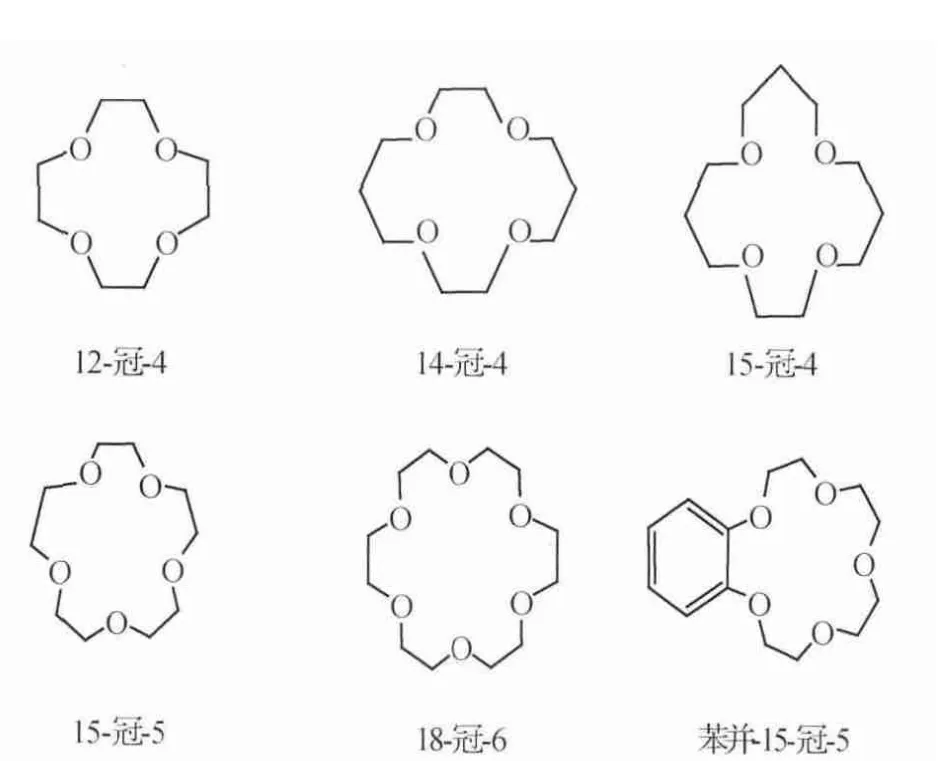
图3 部分冠醚结构
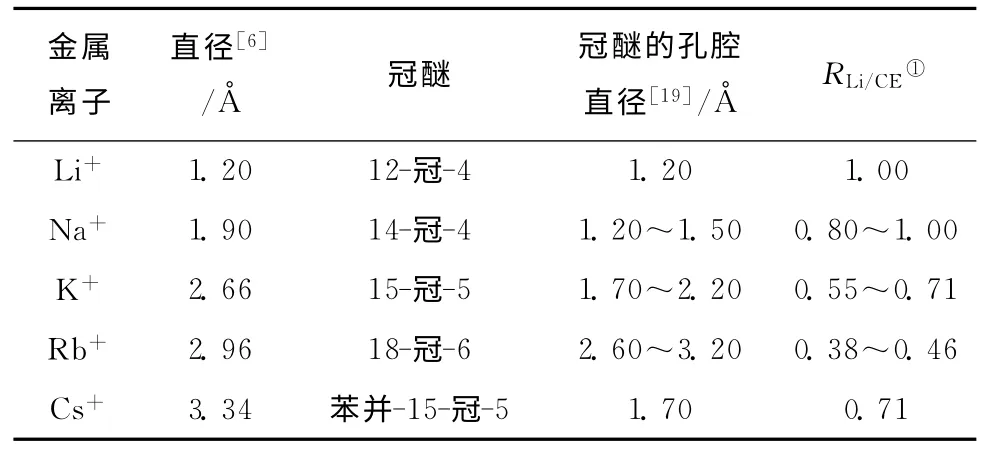
表1 不同碱金属离子直径以及冠醚的孔腔直径比较
刘育等[16]通过液-液萃取的方法,采用萃取率表征冠醚对锂离子的络合能力,考察了不同尺寸的(12~16)-冠-4对 锂 离 子 的 络 合 能 力 变 化(见表2)。作者发现,12-冠-4和13-冠-4的萃取率分别 为2.67%和0.80%,14-冠-4的 萃 取 率 为13.9%。相较于12-冠-4,14-冠-4的锂离子络合能力提升说明14-冠-4的孔径(1.5Å,1Å=0.1nm)比12-冠-4(1.2Å)更有利于对锂离子(1.2Å)的络合。相对于13-冠-4,14-冠-4分子结构更加对称和柔软,这增加了冠醚对锂离子的络合能力。当分子尺寸进一步增大时,15-冠-4和16-冠-4的萃取率分别降低为1.66%和0.29%,这说明过大的环结构降低了给电原子与锂离子的离子-偶极效应,络合作用力降低。Boda等[17]采用从头算和密度泛函理论验证了12-冠-4更有利于络合锂离子的实验结果,他们通过理论计算考查了冠醚-锂络合体系的各种结构,能量及热力学平衡参数,包括各种取代的苯并12-冠-4与锂离子的键能大小顺序,并指出硝基苯较四氯化碳及氯仿更适合作为液液萃取氯化锂的有机相。这一理论计算结果在一定程度上为锂同位素分离萃取提供了参考依据。
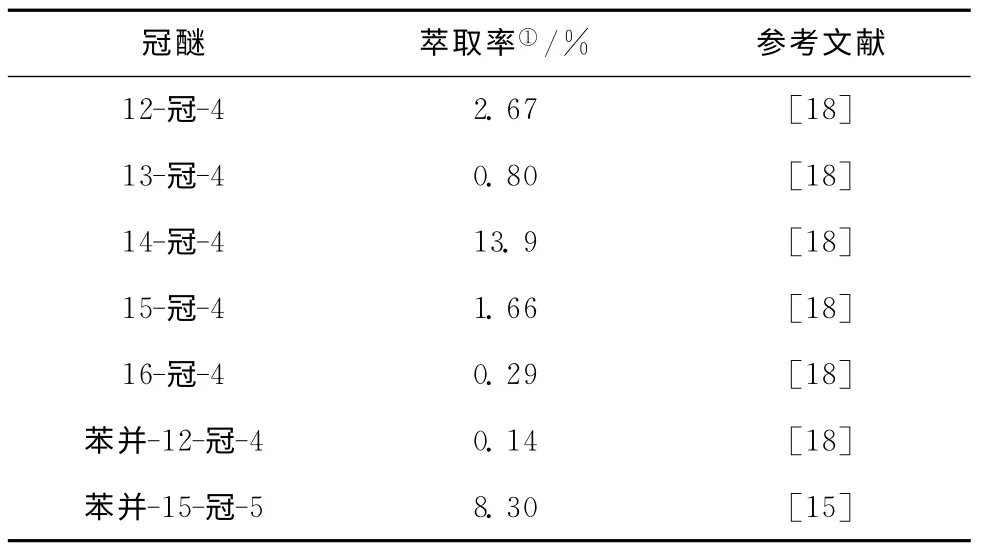
表2 同冠醚结构的锂同位素分离效应
另外,Nishizawa等[14]将苯并15-冠-5溶解于氯仿中制备有机相,将碘化锂水溶液作为水相,在0℃下通过液-液萃取获得苯并15-冠-5的萃取率为8.3%。以上数据表明,14-冠-4和苯并15-冠-5具有对锂离子更好的络合效果。有文献指出[10,15],碱金属阳离子直径与冠醚孔腔尺寸的比值为0.75~0.90时有利于冠醚-离子络合物形成。从表1可以看出,锂离子直径与14-冠-4、苯并5-冠-5的孔径比分别约为0.9和0.7,这与文献[18]报道的尺寸效应规律吻合。
冠醚尺寸的大小对锂同位素分离因子也有影响。Nishizawa等[10]通过液-液萃取考察了不同分子尺寸冠醚的分离因子,并且从热力学的角度进行了影响规律的阐述。作者将12-冠-4、苯并15-冠-5及二环己基18-冠-6分别溶解于氯仿中配制有机相,采用碘化锂水溶液作为水相,通过液-液萃取获得不同冠醚的锂同位素分离热力学参数以及分离因子。表3为不同冠醚的锂同位素分离热力学分析。研究发现,12-冠-4、苯并15-冠-5及二环己基18-冠-6中孔腔尺寸最接近锂离子直径(1.2Å)的冠醚为12-冠-4(1.2Å),苯并15-冠-5(1.7Å)和二环己基18-冠-6的尺寸依次增加(表1)。随着冠醚环尺寸增加,12-冠-4、苯并15-冠-5及二环己基18-冠-6分离锂同位素的焓变和熵变降低,综合的自由能变也降低,对应在0℃下的分离因子分别为1.057、1.042和1.024。

表3 不同尺寸冠醚的锂同位素分离热力学分析[16]
以上结果说明,随着冠醚环尺寸的增加,冠醚对锂离子络合过程中由构象重排导致的焓损耗增加,自由能变降低,导致了分离因子的下降。
综合锂离子络合能力以及分离因子,冠醚孔腔直径接近并略大于1.2Å时有利于提升冠醚的锂同位素分离效应。从热力学的角度讲,冠醚结构的孔腔若匹配锂离子,则冠醚在络合过程中由构象重排带来的焓损失少,过渡态需要活化能小[20],这有利于络合自由能变的增加,从而提升锂同位素分离效应。
2.2 冠醚分子侧基/侧链
侧基对冠醚的离子络合能力具有协同效应,影响锂同位素分离效应。Ungaro等[21]最早系统地考察了具有不同供电吸电能力基团的4取代苯并15-冠-5的钠离子选择络合能力。结果表明,供电基团有利于冠醚络合钠离子,而吸电基团削弱了络合能力。类 似 地,方 胜 强 等[22-23]考 察 了 苯 并15-冠-5,4-甲基苯并15-冠-5以及4-叔丁基苯并15-冠-5的锂同位素分离效应。作者发现,分离因子α并没有随着侧基的变化发生较大改变,但是有机相冠醚络合度P的排序为4-叔丁基苯并15-冠-5>4-甲基苯并15-冠-5>苯并15-冠-5,这与基团供电能力顺序一致,从这一点来看侧基供电将提升锂同位素分离效应。结合图1中12-冠-4的电荷模拟可以发现,当侧基提供额外的电子后将增强冠醚的电荷密度,对应的冠醚中心电负性区也将增强,所以冠醚对锂离子的离子-偶极效应提升,络合力得到增强。若侧基为吸电基团,则冠醚环电荷密度下降,络合能力减弱。
套索醚指具有一个或多个附加侧链的冠醚或者类似大环聚醚的衍生物,是一种典型的通过侧链改变金属离子络合能力的冠醚类型,部分套索醚结构如图4。Hori等[24]系统地考察了带不同侧链的氮杂12-冠-4的锂离子选择络合能力。作者将具有不同侧链的氮杂冠醚分成A型和B型。A型氮杂冠醚的侧链中带有吸电基团(醚、酯和腈基团)包括1a、1b、1c、1d。B型氮杂冠醚的侧链中带有供电基团(氨基或者吡啶基团)包括1e、1f、1g、1h。通过7Li NMR光谱,作者考察了A型和B型氮杂冠醚的锂离子络合物中7Li的化学位移。结果发现,A型 的 络合物中7Li的化学 位 移(Δδ=1.2~1.6)与未带侧链的1a的化学位移(Δδ=1.6)几乎相等,说明A型的侧链未形成锂离子络合的协同作用。而B型的锂络合物中发现7Li化学位移(Δδ=2.1~2.4)明显高于1a。这说明对应的侧链供电原子与锂离子发生离子-偶极作用,7Li离子的电荷密度发生变化,所以出现了明显的化学位移。同时,Hori还对A型和B型氮杂冠醚的锂转移速率进行了考察(见表4)。锂转移速率在文献中体现了冠醚对锂离子的络合速率,转移速率值越大,冠醚络合速率越快。数据显示B型氮杂冠醚中1e、1f的Li+转移速率分别为6.3×107mol/h和8.5×107mol/h,A型的氮杂冠醚中1b、1c的Li+转移速率分别为1.9×107mol/h、0.6×107mol/h,B型氮杂冠醚锂离子络合速率高于A型。基于以上结果,Hori指出带有供电基团的侧链对锂离子提供额外静电作用,有助于锂离子络合力的提升及络合速率的提高。
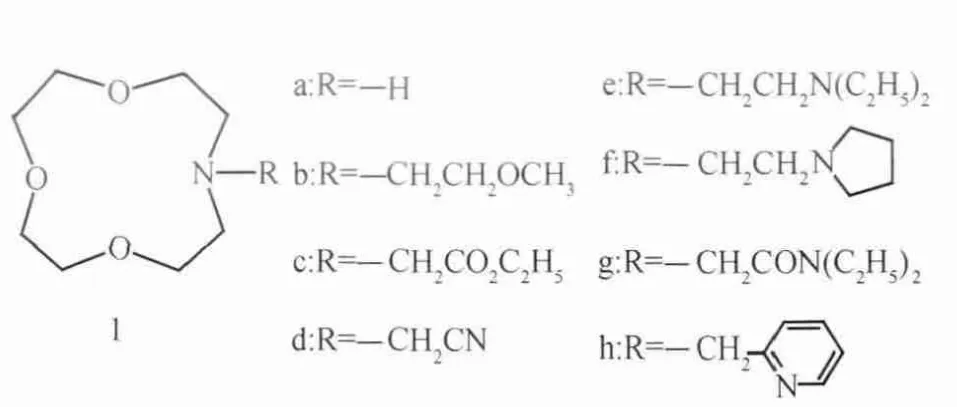
图4 部分套索醚结构[24]
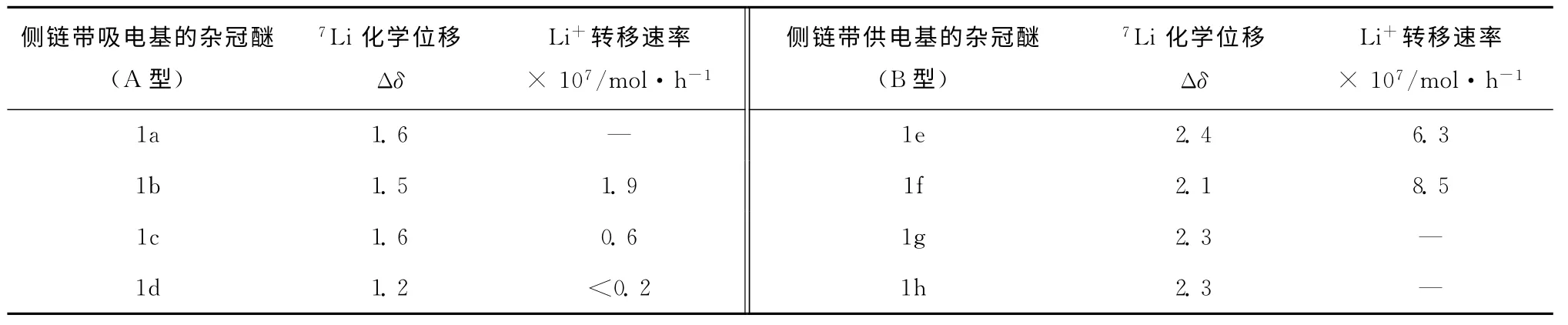
表4 不同类型侧基对Li+络合的影响[24]
另外,在液-液萃取中,侧基还会通过改变冠醚的物化性质(亲水亲油性、溶解度等)提高分离性能。支克正等[25]通过对15-冠-5,环己基15-冠-5及叔丁基环己基15-冠-5考察侧基对冠醚在液-液萃取中锂同位素分离效应的影响。相关实验数据见表5。作者发现,由于冠醚环结构相同,侧基(环己基、叔丁基环己基)没有明显提升冠醚的分离因子。从有机相络合度P来看,15-冠-5由于本身较强的水溶性,在液-液萃取过程中冠醚-锂络合物容易流失到水相,所以有机相络合度仅为1.1%。引入疏水性侧基后,环己基15-冠-5和对叔丁基环己基15-冠-5的有机相络合度分别达到14%和25%,这说明疏水基团降低了冠醚在水相的分配,提高冠醚-锂络合物在有机相的稳定性,有利于锂同位素分离效应的提升。

表5 15-冠-5及其衍生物锂同位素分离效应[25]
综上所述,对于侧基的影响,供电能力强的基团可以增加冠醚电荷密度,提高离子络合力。而带有供电基团(如氨基)的臂式侧链,一方面可以增加冠醚的结构柔韧性,降低络合过渡态能级,加快络合速率;另一方面,长链中供电原子提供的额外离子-偶极作用力也增强了冠醚对锂离子的络合力。除此之外,在液-液萃取体系中,疏水基团增加冠醚在有机相的稳定性,提高萃取效率。
2.3 冠醚环给体原子种类
在软硬酸碱理论中,硬酸配硬碱,软酸配软碱。锂离子属于硬酸,醚键属于硬碱,当冠醚环中的给体原子从氧换成更软的给体(如N,S等),配体对锂离子的络合能力理论上应该是下降的。已有文献指出[26],氮杂冠醚或者硫杂冠醚对软离子Ag+络合能力大大提升,而对碱金属K+的络合能力下降。不过,部分研究显示氮杂冠醚出现了7Li富集树脂相的效果,这使得6Li富集于色谱淋出液的前端,大大提高了高丰度6Li的制备效率[27-28]。
Kim等[11,28-33]对氧氮杂环冠醚树脂体系分离锂同位素进行了大量的研究,涉及的部分杂冠醚结构见图5。表6为杂冠醚树脂材料通过不同色层法获得的分离因子。Kim等[28]首先制备了NTOE杂冠醚树脂,将树脂材料填充进色谱柱(内径0.3cm×高17cm),将少量氯化锂水溶液淋洒于色谱柱上端,之后用0.01mol/L的HCl溶液进行淋洗,获得7Li富集于树脂相,分离因子为1.024。Kim等[27]将N3O3树脂填充色谱柱(内径0.2cm×高35cm),以氯化锂为锂盐,4mol/L的氯化铵水溶液为淋洗液,通过淋洗色谱法获得分离因子为1.028,7Li富集于 树脂相。Kim等[29]将N4O杂 冠醚树脂填充色谱柱(内径0.3cm×高15cm),以氯化锂为锂盐,以0.5mol/L的NH4Cl水溶液为淋洗液,通过淋洗色谱的方法获得分离因子为1.0013,同样也是7Li富集于树脂相,初始淋出液的6Li丰度从7.5%提升到7.8%。以上结果可以看出,当氮杂冠醚树脂倾向于7Li富集时,对应的淋洗色谱中,6Li富集于的淋出液的前段,是一种高效制备6Li的方法。
Betts等[34]曾从理论上研究指出锂同位素分离效应取决于异相间锂键力之差,重同位素7Li富集于锂键强的一相,对于冠醚分离锂同位素体系中,锂离子与冠醚中氧原子的相互作用力相较于溶剂与锂离子的作用力弱,所以6Li富集冠醚相。杂环冠醚出现了7Li富集,这或许是给体原子、尺寸效应以及溶剂、淋洗剂等多种因素共同作用产生的结果,但是目前公开的数据不足,对于机理还未见定论。
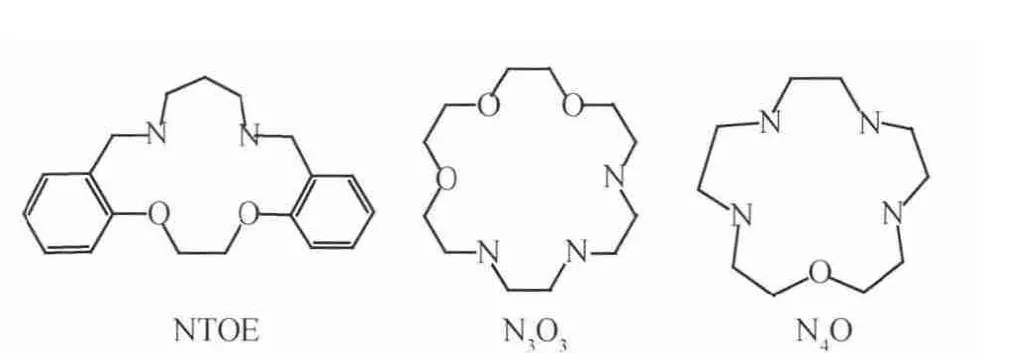
图5 部分杂冠醚结构
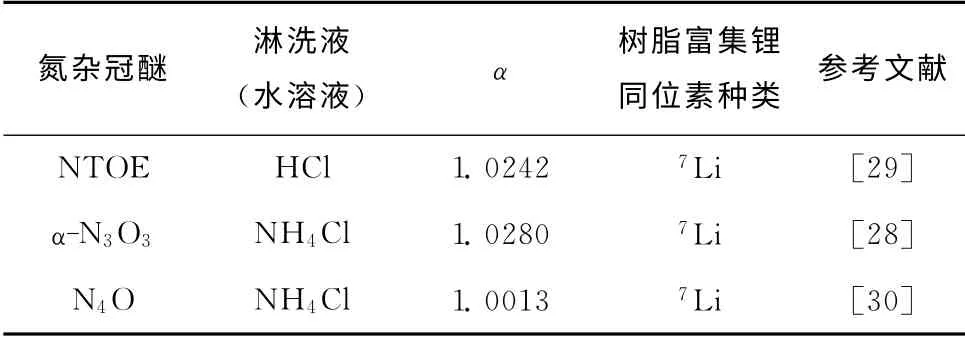
表6 杂冠醚树脂体系锂同位素分离效应
3 化学交换过程中工艺条件对冠醚锂同位素分离效应的影响
通过以上分析可以发现冠醚分子结构对锂同位素分离效应有重要的影响,而化学交换过程的不同工艺条件也可以优化锂同位素分离效应。以下将详细阐述工艺条件(配位阴离子、温度、溶剂种类)对锂同位素分离效应的影响。
3.1 配位阴离子
在冠醚络合锂的过程中,锂离子(Li+),冠醚(L)和阴离子(A-)形成了(LiL)+A-的结构,阴离子的性质对(LiL)+A-的形成以及稳定性都有很重要的影响[14,35-37]。
阴离子的软度和电荷密度影响着冠醚的锂络合力以及分离因子。Nishizawa等[14]将苯并15-冠-5溶解于氯仿制备有机相,将LiCl、LiBr及LiI分别溶解于水中制备水相,通过液-液萃取考察了25℃下3种阴离子对冠醚锂同位素分离效应的影响(数据见表7)。作者发现LiCl、LiBr和LiI 3种锂盐体系下获得有机相冠醚络合度分别为0.1%、0.6%和8.7%,获得的分离因子分别为1.002、1.014和1.026。作者认为冠醚络合度的变化归因于阴离子的软度变化。Cl-、Br-、I-的软度(Em)依次增大,相应为-9.94、-9.22和-8.31[37],这意味着I-更容易向锂离子提供电子,削弱锂离子溶剂化效应,使冠醚受体更好的剔除溶剂分子并结合锂离子,所以LiI体系的络合度最高。分离因子的变化规律主要受到阴离子电荷密度的影响。尽管Cl-、Br-、I-的电荷均为1,但它们的电荷-半径比分别为0.55、0.51及0.45,电荷密度依次降低。当冠醚受体与锂离子发生络合时,配位阴离子电荷密度低,对锂离子的吸引力小,所以冠醚与锂离子间的Li—O配键力变大,同时也增强了冠醚受体与6Li、7Li间的离子-偶极差异,这是分离因子提高的主要因素。
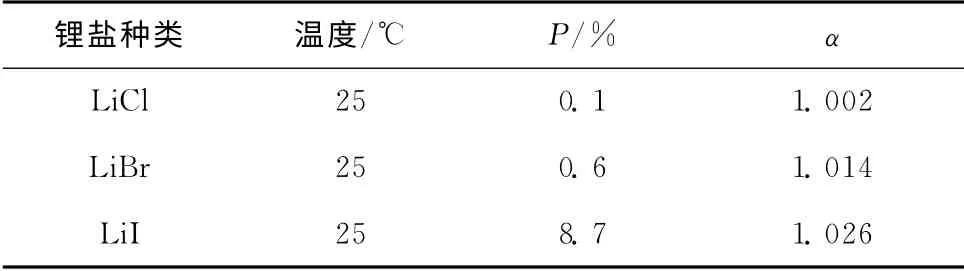
表7 阴离子对苯并15冠5-液-液萃取体系的影响[14]
液-固体系中的阴离子效应与液-液萃取体系是一致的。Nishizawa等[35-36]通过间歇法考察了阴离子对穴醚(2B,2,1)聚合物分离因子的影响。作者将LiCl、LiBr及LiI分别溶解于含有50%(体积比)水的甲醇溶液中,将穴醚(2B,2,1)聚合物投入锂盐溶液中搅拌1h,通过抽滤的方法获得吸附了锂离子的穴醚聚合物。通过分析,作者发现LiCl、LiBr及LiI 3种体系获得的分离因子分别为1.017、1.020和1.023,锂盐的吸附量(mmol/g)分别 为0.111、0.135和0.145。2014年 顾 志 国等[39]采用氧化硅多孔材料固载离子液体和苯并15-冠-5进行锂同位素分离,作者考察了不同阴离子形式锂盐对于萃取分配的影响,不同阴离子锂盐单级分离因子顺序为CF3COOLi>LiSCN>LiI>LiBr>LiCl>LiNO3>CH3COOLi>Li2SO4。以上数据说明阴离子对穴醚聚合物锂同位素分离效应的影响规律是一致的,即阴离子软度的增加提升冠醚受体锂吸附量,阴离子电荷密度降低有利于提升分离因子。
从热力学角度讲,软度大的阴离子有利于锂离子的去溶剂化过程,减少焓损失,而电荷密度小的阴离子增加了冠醚与锂离子的络合焓,提升自由能变。由于相同的热力学规律,阴离子在液-液萃取体系和液-固体系中的锂同位素分离效应是相同的。
3.2 温度
冠醚受体分离锂同位素的过程可以看做恒温恒压自发进行的体系,所以自由能变ΔG°<0。已有的对15-冠-5锂同位素分离效应的研究指出,冠醚分离锂同位素过程的焓变ΔH°和熵变ΔS°为负值,结合热力学方程ΔG°=ΔH°-TΔS°,很容易指出温度(T)越低,自由能变越大,对应获得分离因子将提高。Nishizawa等[36]通过液-液萃取发现苯并15-冠-5的分离因子在1℃、25℃和40℃下的单级分离因子分为1.042、1.026和1.012,单级分离因子随着温度的降低而升高(表8)。总而言之,低温有利于冠醚受体提升锂同位素分离效应。

表8 温度对苯并-15-冠-5锂同位素分离因子的影响[36]
3.3 溶剂
为了形成冠醚-锂络合物,冠醚受体及锂离子必须剔除本身的溶剂化层。从图2可以看出,冠醚和锂离子的去溶剂化将引起焓的损失。所以,无论是液-液萃取体系还是液-固体系,都需要考虑减少溶剂对锂离子以及冠醚分子的溶剂化效应。
在液-液萃取体系中,冠醚小分子需要溶解于有机相,锂盐不可避免需要溶解于水相。锂离子由于较小的半径使得本身电荷密度大,加上水分子较高的电子给体数(表9),所以锂离子水合能力非常强。锂离子在水合作用下形成Li+(H2O)4,水分子削弱锂离子的静电作用,不利于锂同位素分离效应。从表1、表5和表7等数据中可以发现,液-液萃取体系获得的冠醚络合度经常维持在1%左右,这很大一部分原因归结于锂离子的水合作用屏蔽了冠醚和锂离子间的离子-偶极效应。
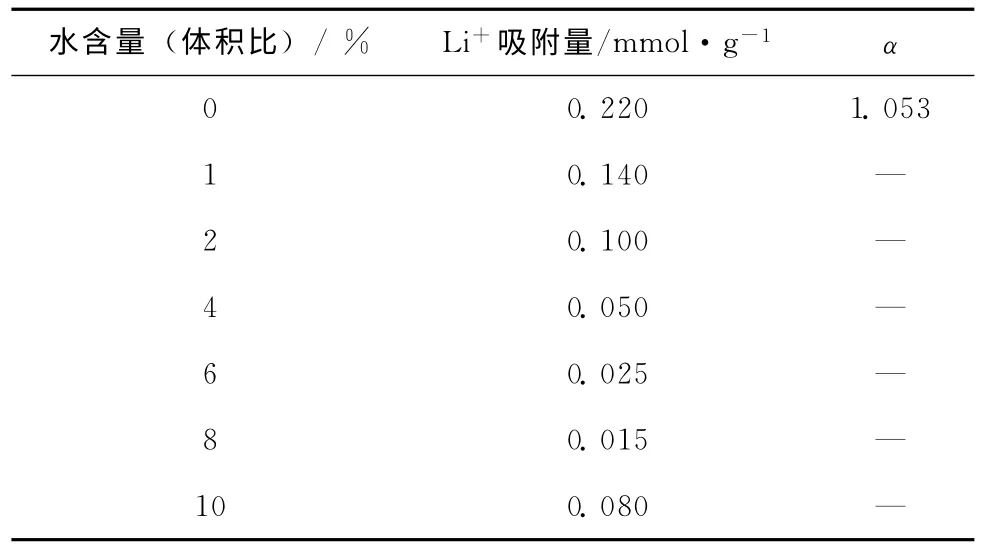
表9 水含量对氨基苯并15冠5树脂的锂同位素分离效应影响[40]
在液-固体系中,由于冠醚结构接枝到了高分子链段中,不再需要溶解于溶剂,所以锂盐的溶剂不再局限于水,这使得液-固体系可以消除水合作用带来的不利影响。Kim等[40]将氨基苯并15冠5接枝到树脂中,通过间歇法考察了液-固体系中水含量对冠醚树脂锂同位素分离效应的影响(表9)。作者配制了不同水含量的乙腈溶液并将LiCl溶解其中,经过液-固萃取后发现,当乙腈中不含水时,锂吸附量达到最高的0.220mmol/g,分离因子达到了1.053,当乙腈水含量增加,冠醚树脂的锂吸附量下降。以上结果说明乙腈中的水含量增加加强了锂离子水合作用,氨基苯并15-冠-5树脂的锂同位素分离效应被削弱。对比表7中的数据,苯并15-冠-5通 过 液-液 萃 取 对LiCl的 分 离 因 子 为1.002,氨 基 苯 并15-冠-5树 脂 在 无 水 的 液-固 体系中分离因子提高到1.053,这说明了无水的液-固体系可以明显提高冠醚受体锂同位素分离效应。
液-固体系中的溶剂选择不再受需要与水互不相容的条件的限制,所以有更多的溶剂种类提供参考。Kim等[41]通 过 间 歇 法 考 察 了 氮 杂 冠 醚DBPDAH螯合树脂在不同电子给体数的溶剂中的锂吸附量以及分离因子(表10)。作者将LiCl溶解于不同电子给体数的溶剂(乙腈、甲醇、二甲基甲酰胺和二甲基亚砜)中,将DBPDAH螯合树脂投入到不同的锂盐溶液进行液-固萃取,结果发现,乙腈体系中获得锂吸附量为0.93mmol/g,分离因子为1.034。随着溶剂电子给体数的增加,树脂锂吸附量和分离因子下降,在二甲基亚砜体系中DBPDAH螯合树脂锂吸附量为0.25,分离因子为1.017。这说明低电子给体数的溶剂可以降低锂离子和冠醚受体的溶剂化效应,使得冠醚受体可以更好的对锂离子进行络合。结合图2,溶剂化效应的减弱有助于主-客体络合中去溶剂化效应的焓损耗,提高锂同位素分离的自由能变,提升锂同位素分离效应。
总而言之,相较于液-液萃取体系,无水的液-固体系消除了锂离子水合作用,有利于提高锂同位素分离效应。在液-固体系中,低电子给体数的溶剂降低溶剂化效应,减少锂离子以及冠醚结构去溶剂化过程的焓损失,提高锂同位素分离自由能变,提升锂同位素分离分离效应。将冠醚固载化形成冠醚聚合物,通过液-固体系分离锂同位素分离是锂同位素的发展趋势。
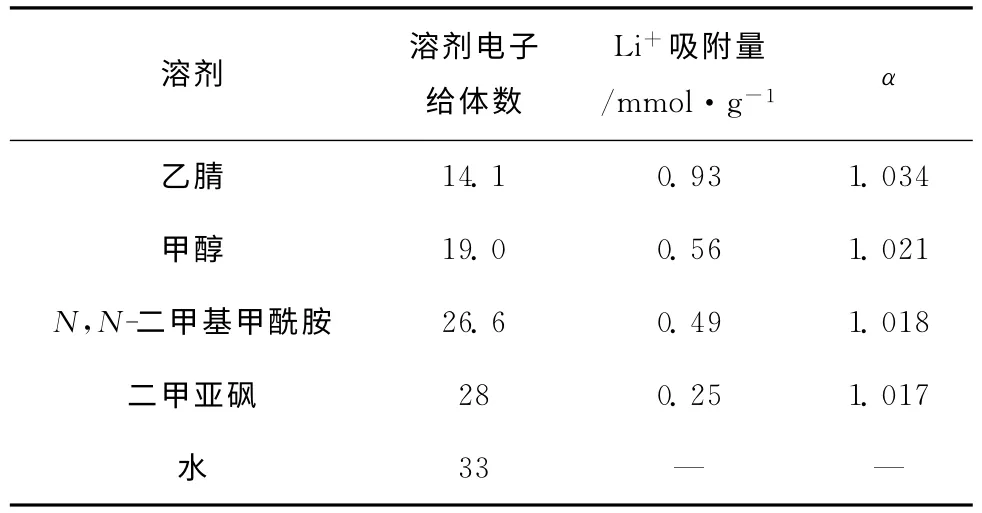
表10 溶剂对锂同位素分离效应的影响[41]
4 总结与展望
冠醚受体通过离子偶极效应实现锂同位素分离,在基于冠醚受体的化学交换法分离锂同位素过程中,冠醚分子的结构(冠醚环大小、给体原子种类、侧链种类)以及不同分离体系(锂盐配位阴离子种类、溶剂种类、化学交换温度)对分离效应有重要的影响。在液-液萃取中,冠醚和锂盐分别溶解于有机相和水相,锂离子水合作用将屏蔽离子-偶极效应。液-固体系中的冠醚结构固载于高分子链段,锂盐的溶剂不再局限于水,形成的无水体系可以提高锂同位素分离效应。此外,冠醚固载于聚合物中,克服了小分子冠醚液-液萃取过程中冠醚流失、无法重复使用以及小分子冠醚毒性等问题[42]。尽管如此,冠醚分离锂同位素至今仍未能实现工业化生产,笔者认为其原因可能在于:一方面,液液萃取体系中冠醚易于流失,难以重复利用;另一方面,高分子固载冠醚分离锂同位素技术还不成熟,停留于研究阶段,且目前的分离量均非常小,还难以达到工业化的要求。因此,进一步研究固载冠醚的液-固体系及其分离锂同位素方法和装置是冠醚分离锂同位素的发展趋势。我国在20世纪90年代末期至2010年间对锂同位素的研究几乎停滞,可喜的是,近几年来继中国原子能研究院、兰州大学等后,中科院上海有机所、上海高等研究院、江南大学及天津工业大学等单位相继加入锂同位素分离领域,开展了固载冠醚分离锂同位素的研究,这将极大促进我国锂同位素分离的研究工作,为我国核能的发展奠定基础。
[1] 顾志国,李在均,杨杰.锂同位素分离[J].化学进展,2011,23(9):1892-1905.
[2] 朱士尧.核聚变原理[M].合肥:中国科学技术大学出版社,1992:5.
[3] 肖啸菴.同位素分离[M].北京:原子能出版社,1999:142.
[4] Thewlis J.Unit-cell dimensions of lithium fluoride made from Li6and Li7[J].ActaCrystallographica,1955,8(1):36-38.
[5] Lewis G N,Macdonald R T.The separation of lithium isotopes[J].J.Am.Chem.Soc.,1936,58:2519-2514.
[6] Pedersen C J.Cyclic polyethers and their complexes with metal salts[J].J.Am.Chem.Soc.,1967,89(26):7017-7036.
[7] Yeong K Y.Complexation studies of crown ethers with alkali mental cations in methanol[D].Malaysia:University Sains Malaysia,2004.
[8] Zollinger D P,Bulten E,Christenhusz A,et al.Computerized conductometric determination of stability constants of complexes of crown ethers with alkali metal salts and with neutral molecules in polar solvents[J].Anal.Chim.Acta,1987,198:207-222.
[9] Jepson B E,DeWitt R.Separation of calcium isotopes with macrocyclic polyether calcium complexes[J].J.Inorg.Nucl.Chem.,1976,38:1175-1177.
[10] Nishizawa K,Takano T,Ikeda I,et al.Extractive separation of lithium isotopes by crown ethers[J].Sep.Sci.Technol.,1988,23(4-5):333-345.
[11] Kim D W,Hong C P,Kim C S,et al.Lithium isotope separation on an ion exchange resin having azacrown ether as an anchor group[J].J.Radioanal.Nucl.Chem.,1997,220(2):229-231.
[12] Davoudi M,Mallah M H,Enrichment of 6Li using dispersive liquid-liquid microextraction as a highly efficient technique[J].Ann.Nucl.Energy,2013,62:499-503.
[13] Steed J W,Atwood J L.Supramolecular Chemistry[M].John Wiley &Sons,Ltd.,2000,94.
[14] Nishizawa K,Ishino S,Watanabe H,et al.Lithium isotope separation by liquid-liquid extraction using benzo-15-crown-5[J].J.Nucl.Sci.Technol.,1984,21(9):694-701.
[15] Rodriguez J D,Lisy J M.Probing ionophore selectivity in argon-tagged hydrated alkali metal ion-crown ether systems[J].J.Am.Chem.Soc.,2011,133,11136-11146.
[16] Liu Y,Inoue Y,Hakushi T.Molecular design of crown ethers.Ⅶ,Syntheses and cation selectivities of unsubstituted 12-to 16-crown-4[J].Bull.Chem.Soc.Jpn.,1990,63(10):3044-3046.
[17] Boda A,Ali S M,Rao H,et al.Ab initio and density functional theoretical design and screening of model crown ether based ligand(host)for extraction of lithium metal ion(guest):Effect of donor and electronic induction[J].J.Mol.Model.,2012,18:3507-3522.
[18] Lamb J,Izatt R,Swain C,et al.A systematic study of the effect of macrocycle ring size and donor atom type on the logK,ΔH,andTΔSof reactions at 25℃in methanol of mono-and divalent cations with crown ethers[J].J.Am.Chem.Soc.,1980,102(2):475-479.
[19] Frensdorf H K.Stability constants of cyclic polyether complexes with univalent cations[J].J.Am.Chem.Soc.,1971,93(3):600-606.
[20] Lehn J M,Sanders J.Supramolecular Chemistry:Concepts and Perspectives[M].Germany:Vch Weinheim,1995:19.
[21] Ungaro R,EL Haj B,Smid J.Substituent effects on the stability of cation complexes of 4'-substituted monobenzo crown ethers[J].J.Am.Chem.Soc.,1976,98(17):5198-5202.
[22] 方胜强,支克正,傅立安,等.4-叔丁基苯并-15-冠-5液-液萃取法分离锂同位素[J].核化学与放射化学,1987,9(3):142-147.
[23] 方胜强,傅立安.4-甲基苯并-15-冠-5分离锂同位素的能力[J].核化学与放射化学,1991,13(2):87-90.
[24] Hori K,Tsukube H.Strategy for designing new Li+ion specific receptors.A combination of theoretical calculations and experimental techniques[J].J.InclusionPhenom.Mol.Recognit.Chem.,1998,32(2):311-329.
[25] 支克正,窦富全,杨坤山,等.冠醚分离锂同位素研究Ⅱ.锂盐-冠醚两相交换体系的锂同位素效应[J].原子能科学技术,1983,3:347-352.
[26] Buschmann H J.The influence of acetonitrile on complex formation of crown ethers containing different donor atoms[J].J.SolutionChem.,1988,17(3):277-286.
[27] Kim D.Enrichment of lithium isotopes by a triazacrown trimerrifield peptide resin[J].J.Radioanal.Nucl.Chem.,2002,253(1):67-71.
[28] Kim D W,Park S P,Kim S J,et al.Separation of lithium isotope by NTOE compound[J].J.Radioanal.Nucl.Chem.,1998,229(1-2):165-168.
[29] Kim D W,Kim B K,Park S R,et al.Separation of lithium isotope by azacrown tetramerrifield peptide resin[J].J.Radioanal.Nucl.Chem.,1998,232(1-2):257-259.
[30] Kim D W,Jeon Y S,Jeong K J,et al.The separation of lithium isotopes with novel azacrown ion exchangers[J].Bull.KoreanChem.Soc.,1995,16(8):683-685.
[31] Kim D W,Kang B M,Jeon B K,et al.Separation of lithium isotopes by elution chromatography with an AB18C6 bonded Merrifield peptide resin[J].J.Radioanal.Nucl.Chem.,2003,256(1):81-85.
[32] Kim D W,Jeon B K,Kang B M,et al.Adsorption and isotope effects by ion exchange with 1-aza-12-crown-4 bonded Merrifield peptide resin[J].J.ColloidInterface Sci.,2003,263:528-532.
[33] Kim D W,Lee N S,Kim C S,et al.Syntheses and separating properties of triazacrown and AM18C6bonded Merrifield peptide resins[J].Eur.Polym.J.,2002,38:2101-2108.
[34] Betts R H,Bron A.Discussion of partial isotope separation by means of solvent extraction[J].J.Sep.Sci.,1977,12(6):635-639.
[35] Nishizawa K,Watanabe H.Intrinsic isotope effect of cryptand(2b,2,1)to li-6and li-7[J].J.Nucl.Sci.Technol.,1986,23(9):843-845.
[36] Nishizawa K,Watanabe H,Ishino S,et al.Lithium isotope separation by cryptand(2B,2,1)polymer[J].J.Nucl.Sci.Technol.,1984,21(2):133-138.
[37] 温永红,杨恩波,傅立安,等.锂盐阴离子和不同侧基的15-冠-5系冠醚对锂盐/冠醚固态配合物远红外位移的影响[J].高等学校化学学报,2007,28(2):224-226.
[38] Klopman G.Chemical reactivity and the concept of chargeand frontier-controlled reactions[J].J.Am.Chem.Soc.,1968,90(2):223-234.
[39] Zhou Wen,Sun Xiao-Li,Gu Lin.et al.A green strategy for lithium isotopes separation by using mesoporous silica materials doped with ionic liquids and benzo-15-crown-5[J].J.RadioanalNucl.Chem.,2014,300:843-852.
[40] Kim D W,Jeon Y S,Eom T Y,et al.Lithium isotope separation on a monobenzo-15-crown-5resin[J].J.Radioanal.Nucl.Chem.,1991,150(2):417-426.
[41] Kim D W,Jeon Y S,Jeong Y K,et al.Lithium isotope separation by chemical exchange with polymer-bound azacrown compounds[J].J.Radioanal.Nucl.Chem.,1995,189(2):219-227.
[42] 李建新,严峰,何本桥,等.一种具有锂同位素分离效应的苯并冠醚接枝聚合物及其制备方法:中国,201210274233.1[P].2012-08-03.
