CCK-8及其受体拮抗剂对吗啡戒断大鼠额叶皮质及海马CREB与pCREB表达的影响*
2014-08-08高平蕊马兴友杨胜昌倪志宇李淑瑾马春玲
高平蕊, 马兴友, 文 迪, 杨胜昌, 于 峰, 倪志宇, 李淑瑾, 马春玲, 丛 斌
(河北医科大学基础医学院法医学系, 河北省法医学重点实验室,河北 石家庄 050017)
八肽胆囊收缩素(cholecystokinin octapeptide,CCK-8)是迄今体内最强的抗阿片肽类物质[1]。有研究证明,CCK 受体与内源性阿片肽受体在中枢神经系统的分布密切相关,参与调节疼痛、焦虑等情绪和记忆过程[2],且在阿片成瘾过程中也发挥着重要作用。阿片成瘾是一种长时程的生物学效应,转录因子cAMP反应元件结合蛋白(cAMP response element binding protein, CREB)在此过程中具有重要的作用,并且磷酸化CREB(pCREB)对下游靶基因转录过程的调控作用可能是吗啡依赖长时程适应性改变的基础之一。本研究通过观察CCK-8及CCK受体拮抗剂对吗啡成瘾大鼠额叶皮质和海马CREB及pCREB的影响,探讨CCK-8 在吗啡成瘾及戒断过程中的分子机制,为CCK-8 在戒毒方面的应用提供相关实验依据。
材 料 和 方 法
1 动物与试剂
成年雄性 Wistar 大鼠 30 只,体质量(200±10) g,由河北省实验动物中心提供(实验动物质量合格证为901014)。盐酸吗啡(沈阳第一制药厂);盐酸纳络酮和CCK-8(Sigma);L-364718和LY-288513(Tocris);CREB、pCREB和β-actin兔抗大鼠单克隆抗体(Cell Signaling);辣根过氧化物酶-山羊抗兔IgG(KPL);化学发光法显色试剂盒(Santa Cruz);链霉卵白素免疫组化染色试剂盒(北京中杉金桥生物技术有限公司)。
2 动物模型的建立及分组
参照文献[3]建立大鼠慢性吗啡依赖及急性催促戒断模型:以剂量递增法连续皮下注射吗啡(10、20、30、40、50 mg·kg-1)5 d,每天2次,间隔12 h(8:00、20:00)。第6天 8:00 皮下注射吗啡 50 mg·kg-1,2 h 后腹腔注射盐酸纳络酮(5 mg·kg-1)进行催促戒断,用改良 Gellert-Holtzman 法[4]评价模型建立是否成功,剔除未成模动物。大鼠随机分组如下:(1)盐水对照组(saline-saline, Sal-Sal):背部皮下注射同等剂量的生理盐水,12只;(2)吗啡依赖组(saline-morphine, Sal-Mor):按照上述方案进行吗啡皮下注射,12只;(3)戒断组(morphine-naloxone, Mor-Nal): 按照上述方案给予吗啡皮下注射后 2 h进行纳络酮催促戒断,12只;(4)慢性药物干预组(CCK-8/L-364718/LY-288513-Mor-Nal):给予吗啡前30 min分别腹腔注射 CCK-8(50 μg·kg-1,n=10)、L-364718(1 mg·kg-1,n=8)、LY-288513(1 mg·kg-1,n=10),余同(3);(5)溶剂对照组(vehicle-Mor-Nal):给予吗啡前 30 min,注射同等体积的溶剂(DMSO : 1,3-丙二醇=1∶4,1 mL·kg-1),余同(3),8只。
3 主要方法
3.1Western blotting 实验结束后应用脑立体定位模具,参照大鼠脑立体定位图谱,急性分离大鼠脑额叶皮质和海马。脑组织按每100 mg组织加入1 mL的组织蛋白裂解液( 50 mmol/L Tris-HCl,pH 7.4,150 mmol/L NaCl,1 mmol/L EGTA,1 mmol/L EDTA,1% NP40,1 mmol/L DTT,1 mmol/L PMSF,1 mg/L aprotinin,1 mg/L pepstatin A,1 mg/L leupeptin) ,用电动匀浆器匀浆30 s,冰浴裂解30 min。4 ℃、12 000 r/min离心20 min 后取上清,用Tiangen公司考马斯亮蓝蛋白定量试剂盒(微板法)测定蛋白浓度,分装后-70 ℃冻存备用。采用SDS-PAGE分离蛋白,用水浴式电转移装置将凝胶上的蛋白质转移到硝酸纤维素膜上,5% 脱脂奶粉封闭1 h,Ⅰ抗4℃孵育过夜(1∶1 000稀释),TTBS洗膜后,加入HRP标记的羊抗兔IgG Ⅱ抗(1∶10 000稀释),37 ℃孵育1 h,化学发光法显色1 min,暗盒中曝光1 min,显影后将X光片用数码相机照相后,用ImageJ图像分析系统对图像进行吸光度扫描,以各组目的蛋白条带吸光度值与内参照蛋白吸光度值计算相对比值,以对照组的比值作为1,其余各组与之相比,再次计算比值作为统计值。
3.2免疫组织化学 实验结束后,用10%水合氯醛麻醉,以0.9%生理盐水行心腔灌注冲洗后,用4%多聚甲醛固定液灌注固定,迅速断头开颅取出全脑,常规制备组织切片、三步法免疫组织化学染色(Ⅰ抗CREB 1∶400、pCREB 1∶50 稀释)。采用Media Cybernetics公司IPP图像分析软件测定阳性神经元平均吸光度值,对CREB和pCREB蛋白表达水平进行半定量分析。
4 统计学处理
数据以均数±标准差(mean±SD)表示,各组均数的比较行单因素方差分析(ANOVA),用最小显著差异(LSD)法进行两两比较,以P<0.05为差异有统计学意义。
结 果
1 吗啡戒断模型的建立
实验中观察到齿颤、湿狗样抖动、腹泻、流泪、流涎、跳跃、体重明显下降等戒断症状,戒断组Gellert-Holtzman评分与依赖组相比明显升高,见图1。

Figure 1. Effects of CCK-8, L-364718 and LY-288513 on the Gellert-Holtzman score of the overall withdrawal seve-rity.Mean±SD. ▲▲P<0.01 vs Mor-Sal group; *P<0.05,**P<0.01 vs Mor-Nal group; #P<0.05 vs L-364718-Mor-Nal group.
2 CCK-8及其受体拮抗剂对吗啡戒断大鼠额叶皮质及海马CREB和pCREB蛋白表达的影响
Western blotting结果显示,额叶皮质和海马CREB蛋白在慢性吗啡作用以及戒断后均无明显变化(P>0.05),见图2A、C;但是pCREB蛋白表达在吗啡依赖后明显增加(P<0.01),在纳洛酮急性催促戒断后进一步升高(P<0.01),见图2B、D。与戒断组(Mor-Nal)相比,CCK-8、L-364718和LY-288513慢性干预对大鼠额叶皮质CREB蛋白表达无明显影响,pCREB蛋白表达明显降低,见图2A、B;海马CREB与pCREB蛋白在L-364718和LY-288513慢性干预后均明显降低,但CCK-8慢性干预对CREB蛋白表达无明显影响,仅pCREB蛋白表达明显降低,见图2C、D。

Figure 2. Effects of CCK-8 and CCK receptor antagonist chronic treatments on the expression of CREB (A,C) and pCREB (B,D) in frontal cortex (A,B) and hippocampus (C,D) of morphine withdrawal rats detected by Western blotting.Mean±SD.n=6 in Sal-Sal, Mor-Sal and Mor-Nal groups; n=5 in CCK-8-Mor-Nal and LY-288513-Mor-Nal groups; n=4 in L-364718-Mor-Nal and Vehicle-Mor-Nal groups. ▲▲P<0.01 vs Sal-Sal group; ■■P<0.01 vs Mor-Sal group; **P<0.01 vs Mor-Nal group.
我们选取了额叶皮质第二运动(M2)区和海马CA1区锥体细胞层神经元作为观察对象,发现正常大鼠额叶皮质M2区神经元胞浆、胞核均表达CREB蛋白,并且慢性吗啡作用后无明显变化(P>0.05)急性纳洛酮催促戒断后CREB蛋白以胞核表达为主,但其表达量无明显变化(P>0.05),见图3;pCREB蛋白则仅在胞核中高表达,慢性吗啡作用后其表达量增加(P<0.01),急性纳洛酮催促戒断后进一步升高(P<0.01),见图4。海马CA1区锥体细胞层神经元中,CREB蛋白在胞浆中高表达,胞核低表达,见图5;pCREB蛋白则在胞核中高表达,见图6。CCK-8、L-364718和LY-288513慢性干预对戒断大鼠额叶皮质M2区和海马CA1区锥体细胞层神经元CREB与pCREB蛋白表达的影响与Western blotting的结果一致。
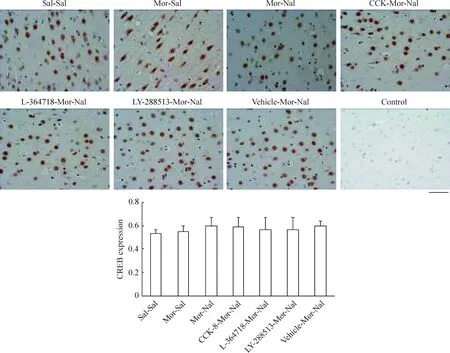
Figure 3. Effects of CCK-8 and CCK receptor antagonist chronic treatments on the expression of CREB in frontal cortex of morphine withdrawal rats detected by immunohistochemistry.Bar=50 μm. Mean±SD.n=6 in Sal-Sal, Mor-Sal and Mor-Nal groups; n=5 in CCK-8-Mor-Nal and LY-288513-Mor-Nal groups; n=4 in L-364718-Mor-Nal and Vehicle-Mor-Nal groups.
讨 论
20世纪80年代,有学者提出脑内可能存在阿片肽的对立面,即抗阿片肽,以维持慢性阿片作用下机体的稳态。胆囊收缩素(cholecystokinin,CCK)是一个具有广泛生物学活性的脑肠肽,它既是胃肠道主要调节激素之一,也作用于中枢神经系统而发挥广泛的生物学作用。CCK-8是迄今体内最强的抗阿片肽类物质,其通过与靶细胞表面的CCK受体结合后发挥多种生物学作用。实验研究表明,CCK受体拮抗剂不但能针对戒断症状进行对症治疗,而且有一定程度的防止复吸作用。Blandine等[3]报道CCK2受体的缺失可以导致内源性阿片肽的释放发生变化。也有研究发现CCK2受体拮抗剂可以有效地减轻慢性吗啡作用后纳络酮催促戒断症状的发生[4],明显抑制吗啡诱导的条件性位置偏爱的形成[5]。然而关于CCK-8减轻吗啡戒断症状的机制尚不完全明确。
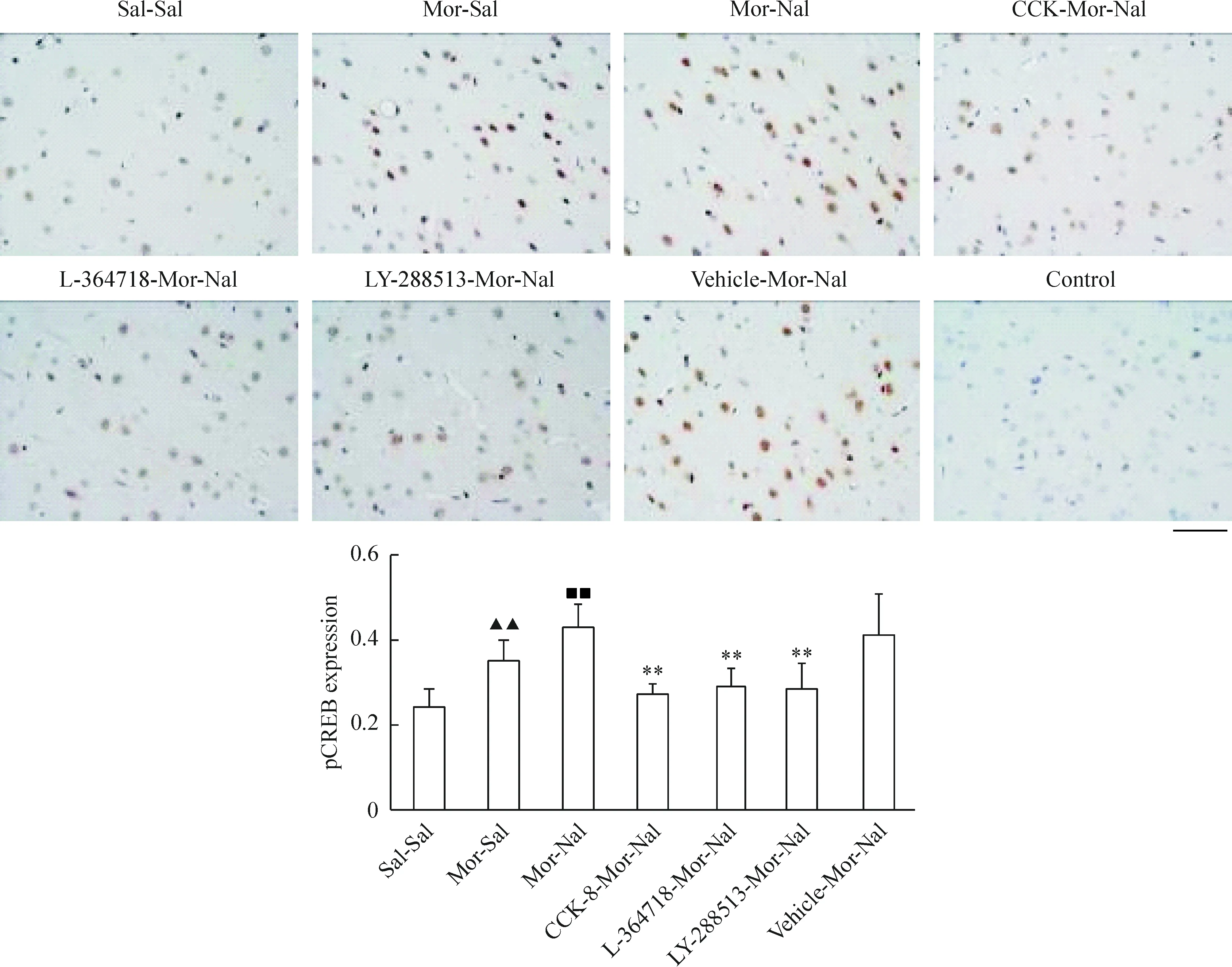
Figure 4. Effects of CCK-8 and CCK receptor antagonist chronic treatments on the expression of pCREB in frontal cortex of morphine withdrawal rats detected by immunohistochemistry.Bar=50 μm.Mean±SD.n=6 in Sal-Sal, Mor-Sal and Mor-Nal groups; n=5 in CCK-8-Mor-Nal and LY-288513-Mor-Nal groups; n=4 in L-364718-Mor-Nal and Vehicle-Mor-Nal groups. ▲▲P<0.01 vs Sal-Sal group; ■■P<0.01 vs Mor-Sal group; **P<0.01 vs Mor-Nal group.
阿片类物质依赖的神经分子生物学机制尚未阐明,其过程涉及到受体、受体后的信号转导,基因的转录、翻译和调控,效应子的表达及生物效应的产生等多个方面。已知急性吗啡可以降低细胞内cAMP水平。然而,随着吗啡持续处理,cAMP水平逐渐恢复到正常。再用阿片受体拮抗剂处理,cAMP水平就增加到远远超过正常值。cAMP的上调可促进PKA活性的增加,进而促进CREB的转录因子特异性丝氨酸残端磷酸化。CREB属于碱性氨基酸亮氨酸拉链转录因子家族,它具有转录活性的二聚体和无转录活性的单体。Ser133是CREB激活的关键位点,磷酸化后的CREB 形成同源二聚体或与CREB/ATF 家族的其它成员形成异二聚体,使CREB活化[6]。目前普遍认为,磷酸化激活的CREB形成二聚体与cAMP反应元件(cAMP response element, CRE)结合,引起一组靶基因表达上调,导致多种蛋白产物持续增多,参与了应激、认知等功能的调节[7-8]。另外,CREB诱导的蛋白表达还可引起神经元可塑性和适应性改变,这种适应性的改变是慢性吗啡依赖和耐受的共同环节,也是形成慢性吗啡依赖和戒断的重要机制之一[9-10]。CREB第133位丝氨酸磷酸化是其调控基因表达的基本条件,CREB 磷酸化的水平与细胞膜的去极化能力有关, 介导细胞膜的去极化过程, 诱导c-fos基因的表达。磷酸化CREB对下游靶基因转录过程的调控作用可能是吗啡依赖长时程适应性改变的基础之一,CREB在药物依赖的形成及表达过程中起着承上启下的作用。
吗啡成瘾的形成是由多个脑区共同参与的结果。额叶皮质参与了中脑-皮层多巴胺能系统,涉及高级认知功能与动机功能。海马作为边缘结构则广泛地参与了学习与记忆过程,尤其是长期记忆过程的形成,并且在吗啡成瘾记忆形成过程中起重要作用。慢性阿片处理能减弱海马CA1区突触的长时程增强,改变海马的突触可塑性[11]。研究表明额叶皮质、海马等脑区不仅在成瘾记忆方面发挥着重要作用,同时也参与躯体戒断症状的形成[12-13]。

Figure 5. Effects of CCK-8 and CCK receptor antagonist chronic treatments on the expression of CREB in hippocampus of morphine withdrawal rats detected by immunohistochemistry.Bar=50 μm.Mean±SD.n=6 in Sal-Sal, Mor-Sal and Mor-Nal groups; n=5 in CCK-8-Mor-Nal and LY-288513-Mor-Nal groups; n=4 in L-364718-Mor-Nal and Vehicle-Mor-Nal groups.**P<0.01 vs Mor-Nal group.
在Western blotting实验结果中,我们发现慢性吗啡作用后,大鼠额叶皮质和海马CREB蛋白表达无明显变化,但pCREB蛋白表达明显增加。纳洛酮急性催促戒断后,CREB蛋白表达仍未发生明显变化,pCREB蛋白表达进一步升高。与戒断组相比,CCK-8、L-364718和LY-288513慢性干预对大鼠额叶皮质CREB蛋白表达无明显影响,pCREB蛋白表达明显降低;L-364718和LY-288513急性干预对大鼠额叶皮质CREB/pCREB蛋白表达均无明显影响。海马CREB和pCREB蛋白表达在L-364718和LY-288513慢性干预后均明显降低,但CCK-8慢性干预及L-364718、LY-288513急性干预对CREB蛋白表达无明显影响,仅pCREB蛋白表达明显降低。CCK-8急性干预对戒断大鼠额叶皮质、海马CREB与pCREB蛋白表达均无明显影响。免疫组织化学结果发现正常大鼠额叶皮质M2区神经元胞浆、胞核均表达CREB蛋白,并且慢性吗啡作用后无明显变化;急性纳洛酮催促戒断后CREB蛋白以胞核表达为主,但其表达量无明显变化。pCREB蛋白则在胞核中高表达,慢性吗啡作用后其表达量增加,急性纳洛酮催促戒断后进一步升高。海马CA1区锥体细胞层神经元中,CREB蛋白在胞浆中高表达,而胞核低表达;pCREB蛋白则在胞核中高表达。CCK-8、L-364718和LY-288513慢性干预对戒断大鼠额叶皮质M2区和海马CA1区锥体细胞层神经元CREB与pCREB蛋白表达的影响与Western blotting实验中的结果一致。
分析所得到的实验结果,我们考虑给予CCK受体拮抗剂后,拮抗了内源性CCK的作用,使CCK的生理效应丧失,减轻了吗啡戒断症状,并使受体后途径的相关指标发生了相应变化。CCK受体包括CCK1和CCK2受体,分别与Gq和Gq/Gs偶联。CCK1受体通过IP3-Ca2+-CaMK-CREB以及DAG-PKC-CREB信号通路,CCK2受体通过cAMP-PKA-CREB以及DAG-PKC-CREB信号通路调节下游基因表达,参与体内多种生理功能的调节。这提示CCK1和CCK2受体拮抗剂均可通过各自的信号转导通路降低吗啡戒断大鼠额叶皮质和海马内CREB磷酸化的水平,进而减轻吗啡戒断症状。而CCK-8慢性干预却减轻了纳洛酮引起的急性催促戒断症状,与CCK受体拮抗剂的作用一致。本文认为,这与CCK-8的效应浓度有密切关系[14-16],并推测在注射吗啡前外源性给予高浓度CCK-8慢性干预时,发挥的是药理效应,其可能通过抑制外源性阿片物质和阿片受体的结合,或者影响外源性阿片物质所产生的后效应而发挥作用;另一方面,高浓度的CCK还有可能通过直接激活阿片受体发挥作用[17],其具体机制还需进一步探讨。

Figure 6. Effects of CCK-8 and CCK receptor antagonist chronic treatments on the expression of pCREB in hippocampus of morphine withdrawal rats detected by immunohistochemistry. Bar=50 μm. Mean±SD.n=6 in Sal-Sal, Mor-Sal and Mor-Nal groups; n=5 in CCK-8-Mor-Nal and LY-288513-Mor-Nal groups; n=4 in L-364718-Mor-Nal and Vehicle-Mor-Nal groups..▲▲P<0.01 vs Sal-Sal group; ■■P<0.01 vs Mor-Sal group; **P<0.01 vs Mor-Nal group.
总之,CCK在吗啡成瘾及戒断过程中发挥了重要作用,并通过调节受体后相关转录调控信号蛋白CREB的表达及其磷酸化过程而发挥此作用,为戒毒提供了一种新的思路。
[参 考 文 献]
[1] Faris PL, Komisaruk R, Watkins LR, et al. Evidence for the neuropeptide cholecystokinin as an antagonist of opiate analgesia[J]. Science, 1983, 219(4582): 310-312.
[2] Hebb AL,Poulin JF, Roach SP, et al. Cholecystokinin and endogenous opioid peptides: interactive influence on pain, cognition, and emotion[J]. Prog Neuropsychopharmacol Biol Psychiatry, 2005, 29(8):1225-1238.
[3] Blandine P,Franc B,Axelle S,et al.Deletion of CCK2 receptor in mice results in an upregulation of the endogenous opioid system[J]. J Neurosci, 2002, 22(5):2005-2011.
[4] Noble F, Wank A, Crawley JN, et al. International Union of Pharmacology. XXI. Structure, distribution and functions of cholecystokinin receptors[J]. Pharmacol Rev, 1999, 51(4):745-781.
[5] Mitchell JM, Bergren LJ, Chen KS, et al. Cholecystokinin is necessary for the expression of morphine conditioned place preference[J]. Pharmacol Biochem Behav, 2006, 85(4):787-795.
[6] Chao J, Nestler EJ. Molecular neurobiology of drug addiction[J]. Annu Rev Med, 2004, 55:113-132.
[7] 于 琦, 金光亮. 三种复方对慢性应激模型大鼠海马CREB、BDNF基因表达的影响[J]. 中国病理生理杂志, 2009, 25(3):591-594.
[8] 蔡晓红, 张存雪, 周永海, 等. 慢性间歇低氧对幼鼠认知及相关脑区CREB的影响[J]. 中国病理生理杂志, 2010, 26(5):895-900.
[9] Goldstein A,Tachibana S,Lowney LI,et al. Dynorphin-(1-13),an extraordinary potent opioid peptide[J]. Proc Natl Acad Sci U S A, 1979, 76(12):6666-6670.
[10] 马春玲, 杨胜昌, 文 迪, 等. CCK-8对吗啡戒断大鼠额叶皮质、海马CREB和pCREB的影响[J]. 中国病理生理杂志, 2010, 26(10):2052.
[11] Pu L, Bao GB, Xu NJ, et al. Hippocampal long term potentiation is reduced by chronic opiate treatment and can be restored by re-exposure to opiates [J]. J Neurosci, 2002, 22(5):1914-1921.
[12] Ammon S, Mayer P, Riechert U, et al. Microarray analysis of genes expressed in the frontal cortex of rats chronically treated with morphine and after naloxone precipitated withdrawal[J]. Brain Res Mol Brain Res, 2003, 112(1-2):113-125.
[13] Fan GH, Wang LE, Qiu HC, et al. Inhibition of calcium/calmodulin-dependent protein kinase-II in rat hippocampus attenuates morphine tolerance and dependence[J]. Mol Pharmacol, 1999, 56(1):39-45.
[14] Doi T, Jurna I. Analgesic effect of intrathecal morphine demonstrated in ascending nociceptive activity in the rat spinal cord and ineffectiveness of caerulein and cholecystokinin octapeptide[J]. Brain Res, 1982, 234(2):399-407.
[15] Barbaz BS, Hall NR, Liebman JM.Antagonism of morphine analgesia by CCK-8-S does not extend to all assays nor all opiate analgesics[J]. Peptides, 1988, 9(6):1295-1300.
[16] Rezayat M, Azizi N, Zarrindast MR. On the mechanism(s) of cholecystokinin (CCK): receptor stimulation attenuates morphine dependence in mice[J]. Pharmacol Toxicol, 1997, 81(3):124-129.
[17] Romanelli L, Amico MC, Mattioli F, et al. Interactions between cholecystokinin and opioids in the isolated guinea-pig ileum[J]. Br J Pharmacol, 1999, 127(4): 909-918.
