负载型杂多酸催化甲苯异丙基化反应
2014-07-24冯锡兰彭慧慧柳云骐刘晨光戴昉纳刘春英
冯锡兰,彭慧慧,柳云骐,刘晨光,戴昉纳,刘春英
(中国石油大学(华东)CNPC 催化重点实验室,山东 青岛 266580)
贫邻位异丙基甲苯(IPT)作为重要的化工产品和用途广泛的有机合成中间体,其合成技术和工艺一直备受关注。工业上通常是以甲苯和丙烯或异丙醇为原料,在酸催化剂作用下通过Friedel-Crafts 烷基化反应来实现。传统的工业催化剂主要采用三氯化铝和磷酸/硅藻土催化剂,二者均存在设备腐蚀和污染问题,且产物的对位异构体选择性低。针对这种状况,分子筛催化剂以其可调变的酸性、独特的择形性以及环境友好的特点成为催化甲苯异丙基化反应的研究热点。1972年Mobil 公司首次在甲苯的异丙基化反应中使用了ZSM-5 分子筛,以后陆续又有HM、HY、Hβ、HZSM-12 等系列分子筛应用于甲苯烷基化反应。研究者通过不同方法对分子筛进行改性,调变其酸性和孔道结构,获得了较优的活性和选择性,并已有工业化应用[1-3],但依然有转化率不高、催化剂易结焦失活等问题。因此,具有优良酸催化性能的杂多酸化合物进入人们的视线。
杂多酸化合物具有独特的酸性,是酸强度较为均一的质子酸,它同时还具有氧化还原性以及独特的“假液相”行为,这一特征使催化反应过程不只局限于催化剂的表面进行,还能进入催化剂的体相,因而杂多酸具有优异的催化活性和选择性,在氧化、酯化、傅克烷基化、环氧化物开环、脱水等反应中均有广泛的应用[4-8]。但由于杂多酸的比表面积非常小(<10m2/g),且在应用于催化反应中存在着回收困难的问题,因此杂多酸的固载化及其催化应用成为催化领域的研究热点[9-13]。负载型杂多酸通过杂多酸在载体上的分散增大了活性组分的比表面积,从而使活性组分在催化过程中得到充分利用,且最后催化剂与产物易于分离和回收。负载型杂多酸的结构、酸性、氧化-还原性会受到载体材料性质及负载方法的影响,这是杂多酸固载化需要研究解决的问题。目前应用于负载杂多酸的载体主要有:活性炭、SiO2、介孔分子筛、过渡金属氧化物以及离子交换树脂等[14-17];负载方法常见的有吸附法、浸渍法、离子交换法、共沉淀法和溶胶-凝胶法等[18],其中浸渍法因其工艺简单、负载量可调范围广、杂多酸活性易于保持等优点获得普遍青睐。但为解决活性组分易溶脱的不足,必须选择比表面积大、与杂多酸吸附作用强的载体。凌云等[19]以多孔硅胶为载体,采用浸渍法制备了负载型杂多酸催化剂,并将其应用于甲苯和丙烯烷基化生产间异丙基甲苯的反应,结果表明,催化剂在反应中保持了很好的活性和稳定性,具有工业化应用前景。但实验着重于考察反应工艺条件的影响,未对催化剂制备的影响因素作深入探究。
本文采用浸渍法将磷钨酸(PWH)负载于二氧化硅载体上制备负载型杂多酸催化剂(PWH/SiO2),并对其应用于催化甲苯与异丙醇烷基化反应进行了较为系统的研究,以异丙醇转化率、烷基化反应选择性及o-IPT 选择性为目标,考察了PWH 负载量和焙烧温度对催化剂结构和性能的影响。
1 实验部分
1.1 原料和试剂
甲苯(C7H8,分析纯,西陇化工股份有限公司),异丙醇(C3H8O,分析纯,西陇化工股份有限公司),磷钨酸(PWH,分析纯,国药集团化学试剂有限公司),大孔硅胶(工业品)。
1.2 催化剂的制备
以磷钨酸作为催化剂活性组分,以SiO2作为载体,制备负载型杂多酸催化剂。
(1)载体大孔硅胶的预处理 硅胶用5%硝酸浸泡4h,然后用蒸馏水洗至中性,洗后的硅胶于120℃下干燥10h,其后于550℃下焙烧4h。将处理过的硅胶研磨,筛选20~40 目硅胶备用。
(2)负载型杂多酸的制备 采用等体积浸渍法制备负载型杂多酸催化剂。按照所需的负载量将一定量的磷钨酸溶于一定量的去离子水中,得到浸渍液。取适当体积的浸渍液,加入所需的载体(浸渍液的量恰好为所需载体的饱和吸水量),室温下浸渍24h,浸渍完成后,将其置于105℃下干燥4h,最后于马弗炉中一定温度下焙烧3h。
1.3 催化剂的表征
1.3.1 X 射线衍射(XRD,表征催化剂的晶相结构)
样品的晶相结构表征在荷兰帕纳科公司新型X′Pert Pro MPD X 射线衍射仪上进行,测试条件为:Cu 靶Kα辐射源,管压40kV,管流40mA。扫描范围2θ 为5°~75°。
1.3.2 吡啶吸附红外(Py-IR,表征催化剂的表面 酸性)
样品酸类型在美国 Thermo Nicolet 公司NEXUS 型FT-IR 红外光谱仪上采用吡啶吸附红外光谱法表征。样品在干燥箱中120℃下干燥8h,然后在马弗炉中200℃焙烧3h,将预处理好的样品于真空干燥器中吸附吡啶12h,样品吸附饱和后在真空干燥箱中120℃下脱附1h。以未吸附吡啶的样品做参比,进行红外漫反射。
1.3.3 低温N2吸附容量法(测定催化剂的孔径、孔容和比表面积)
用静态低温氮气吸附容量法表征催化剂的孔径、孔容和比表面积,在美国 Micromeritics ASAP2010 型自动吸附仪上完成。样品在200℃下抽真空至7~10mmHg 柱,处理完毕后,回充N2至常压,以BET 方法测算样品的比表面积,以BJH等效圆柱模型估算样品的孔体积及孔径分布。
1.4 催化剂活性的评价
1.4.1 实验装置
催化剂活性评价在高压流动微反装置上进行,实验装置见图1。载气为氢气。实验时首先将按比例混合好的原料置换整个管路,待管路中的气体完全排净后,将系统压力调至2MPa,并将催化剂床层温度程序升温至反应温度,然后打开原料泵进样。反应后的液体产物定期取样,供分析和检测用。
1.4.2 产物的分析方法
产物组成及定量分析在Agilent GC7890 气相色谱仪上进行,内标法定量。测试条件为:FID 检测器,HP-PONA Methyl Siloxane 型色谱柱,规格为 50m × 200μm × 0.5μm,柱箱温度为程序升温,汽化室温度为250℃,检测室温度为300℃,载气流量为35mL/min,助燃气流量为350mL/min,进样量为0.5μL。
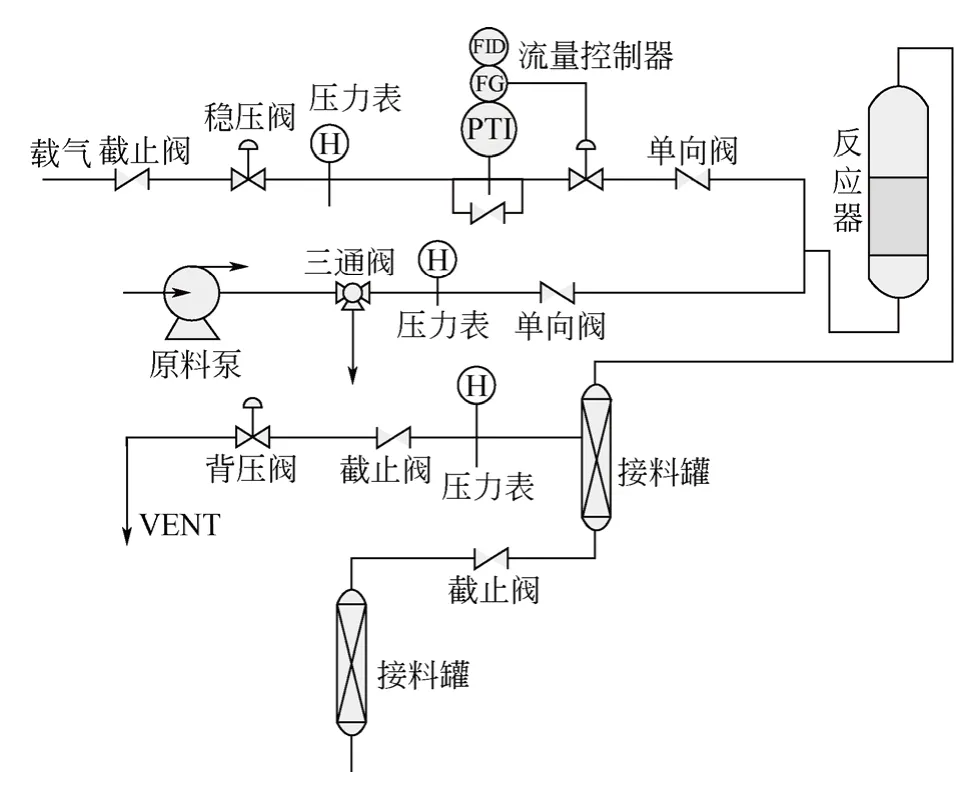
图1 催化剂性能评价装置
1.4.3 内标法色谱数据计算
选择异丙苯为内标物,通过内标法进行定量 计算。
(1)产物中各组分含量的计算 准确称量试样,加入一定质量的异丙苯作为内标物,进行色谱分析。其中,物质的质量之比等于其峰面积之比与校正因子的乘积,即式(1)所示。
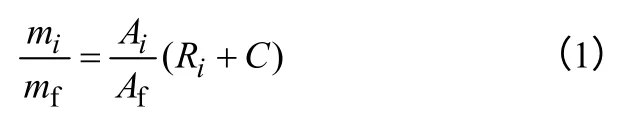
式中,mi、mf分别为试样中某组分的质量、异丙苯的质量;Ai、Af为组分、异丙苯在色谱图中的峰面积;Ri为该组分的校正因子。
(2)异丙醇转化率的计算 已知试样质量和进料时甲苯及异丙醇的摩尔比,可以计算得到相同质量下原料中异丙醇的总质量。因此可由式(2)求得异丙醇的转化率。
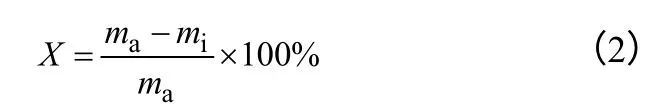
式中,X 为异丙醇的转化率;ma为原料中异丙醇的质量;mi为产物中异丙醇的质量。
(3)烷基化反应选择性的计算 已知异丙基甲苯各异构体的峰面积,根据异丙基甲苯各异构体校正因子即可计算得到各异构体的质量。烷基化反应的选择性可通过式(3)进行计算。
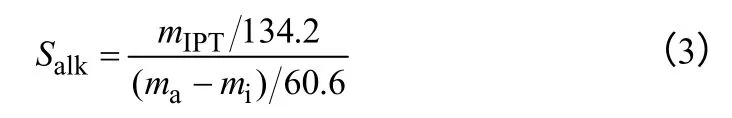
式中,Salk为烷基化反应的选择性;mIPT为试样中异丙基甲苯各异构体的质量之和;ma-mi为异丙醇发生转化的质量。
(4)异丙基甲苯各异构体选择性的计算 对-异丙基甲苯、间-异丙基甲苯和邻-异丙基甲苯的选择性分别由式(4)、式(5)和式(6)计算得到。
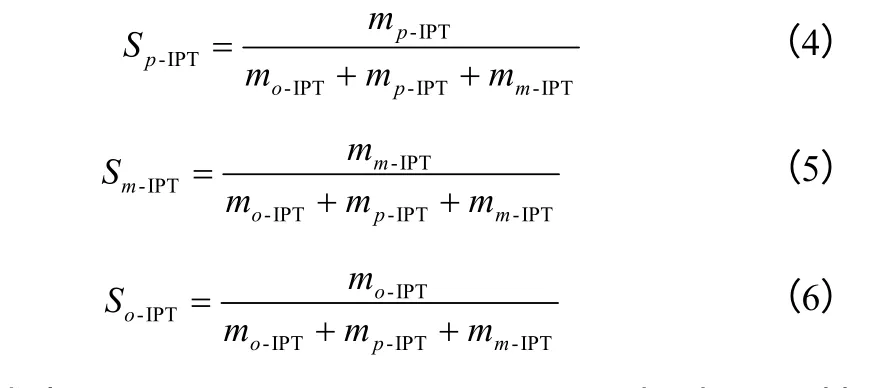
式中,Sp-IPT、Sm-IPT、So-IPT分别为对-异丙基甲苯(p-IPT)、间-异丙基甲苯(m-IPT)和邻-异丙基甲苯(o-IPT)的选择性;mp-IPT、mm-IPT及mo-IPT分别为对、间和邻-异丙基甲苯的质量。
2 结果与讨论
2.1 PWH/SiO2 催化剂结构分析
杂多酸与载体之间存在着相互作用,因此杂多酸负载后结构会有所变化,而杂多酸结构的变化对催化剂的催化活性起着决定性作用,因此对杂多酸负载后的结构进行研究非常重要[20]。对PWH、载体SiO2及负载型杂多酸PWH/SiO2进行IR 表征,结果见图2。
由图2 可知,Keggin 结构的PWH 在1080cm-1、982cm-1、897cm-1、804cm-1有4 个吸收峰,分别归属于P—O 键、W—Ot(端氧)键、W—Oe(八面体中共边氧)键以及W—Oc—W(3 个WO6八面体的共角氧)键的伸缩振动峰,与文献[21]报道Keggin结构的IR 吸收峰一致。负载型PWH/SiO2中磷钨酸的4 个特征吸收峰依然存在,说明磷钨酸在负载后其基本结构未发生变化,但磷钨酸在1080cm-1处的特征峰与载体SiO2在1100cm-1处的特征峰重叠为1112cm-1处的宽峰,并发生了一定程度的红移,这表明磷钨酸与载体SiO2之间存在相互作用。

图2 PWH、载体SiO2 及负载杂多酸PWH/SiO2 的IR 谱图 (200℃焙烧)
Lefebvre[22]研究了PWH 在SiO2上的吸附作用,通过固体核磁研究确认PWH 的H+与SiO2表面的羟基作用能形成(≡SiOH2+)(H2PW12O40-)。
2.2 不同负载量对PWH/SiO2 催化剂性质的影响
2.2.1 催化剂的酸性
采用吡啶吸附红外对样品的酸性进行表征,其中1450cm-1处的吸收峰代表L 酸,1540cm-1处的吸收峰代表B 酸。
图3 所示为载体SiO2、PWH 及负载量分别为30%、40%和50%杂多酸(PWH/SiO2)催化剂(200℃焙烧)的吡啶吸附红外谱图。由图3 可见,SiO2的谱线图在1540cm-1附近未见明显的谱峰,说明硅胶不具有B 酸中心,而在1450cm-1处有强的L 酸峰。PWH 谱线则显示出强的B 酸峰和较弱的L 酸峰。负载杂多酸(PWH/SiO2)催化剂在1540cm-1处出现不同强度(负载量不同)的B 酸峰,并且随负载量的增加,B 酸强度也随之增大,同时其1450cm-1处的L 酸峰均强于SiO2和PWH,为二者的叠加。此外,在1490cm-1和1635cm-1处都可明显看出负载后的催化剂保留了磷钨酸的特征峰。由此推断,负载之后的磷钨酸的基本结构并没有发生变化,确证了Keggin 结构不会因负载量的改变而变化。
2.2.2 活性组分在载体上的分散
对于负载型催化剂,活性组分在载体上的分散情况是影响催化活性的一个重要因素。活性组分的分散程度可以由XRD 谱图所提供的信息获得。图4所示为载体SiO2、PWH 及负载量分别为30%、40%和50%杂多酸催化剂(200℃焙烧)的XRD 谱图。
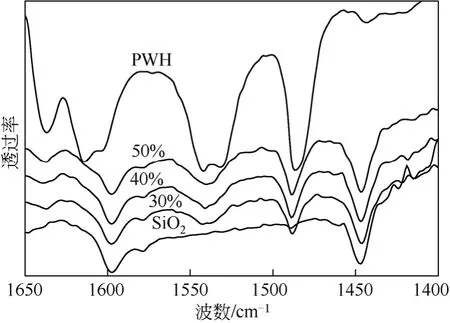
图3 200℃焙烧不同负载量PWH/SiO2的吡啶吸附红外光谱
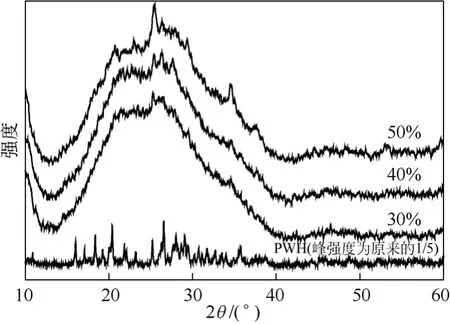
图4 PWH 及200℃焙烧不同负载量的PWH/SiO2的XRD 图
由图4 可以看出,随着负载量的不断增加,磷钨酸的微晶衍射峰逐渐增强和增多。当磷钨酸负载量为30%时,其XRD 谱图与SiO2的谱图非常接近,基本看不到磷钨酸的特征峰,说明在该负载量下,磷钨酸能够较为均匀地分散在载体SiO2表面。当负 载量为40%时,出现了磷钨酸特征峰,而当负载量达到50%时,磷钨酸的特征衍射峰已非常明显。由此可见,采用SiO2为载体,磷钨酸首先以单层形式分布在SiO2的表面,随磷钨酸负载量的增加,超过了磷钨酸在载体表面的分散阈值而逐渐聚集,形成了磷钨酸微晶。
2.2.3 催化剂比表面积与孔结构
固体催化剂的比表面和孔结构与其催化性能有着密切关系,通常采用N2吸附脱附法进行表征。表1 所示为不同负载量下催化剂的比表面积与孔结构。由表1 中数据可以看出,随着磷钨酸负载量的不断增加,催化剂的比表面积和孔容都逐渐下降,而催化剂的孔径则呈现先减小后增大的趋势。载体SiO2的比表面积为346.13m2/g,磷钨酸负载量为30%、40%和50% PWH/SiO2催化剂的比表面积分别为265.46m2/g、252.48m2/g、230.84m2/g,不难看出,负载磷钨酸之后比表面积下降明显,说明磷钨酸占据了载体的表面。而负载量的变化并没有使比表面积发生较大的改变,催化剂比表面积的下降相对缓慢,分析其原因可能是磷钨酸在载体表面发生了聚集,使得铺展不再均匀。随着负载量的增加,催化剂的孔径先减后增,则是因为当负载量增大至一定程度时,活性组分在载体表面的聚集程度增大,会堵塞较小的孔所致。
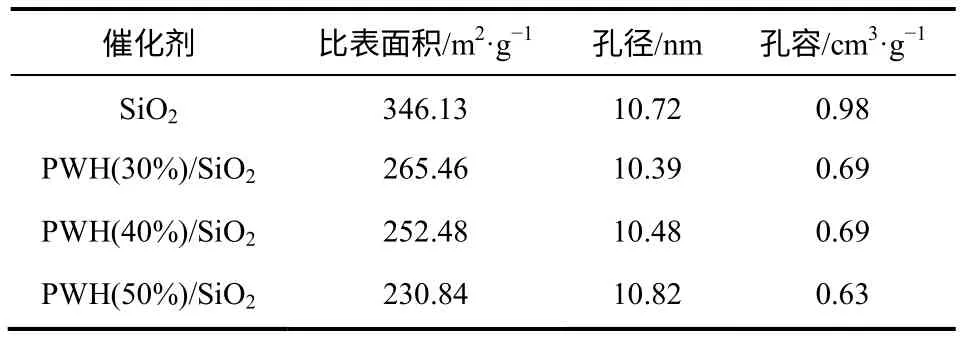
表1 不同负载量PWH/SiO2 的比表面积与孔结构
2.2.4 不同负载量PWH/SiO2的催化性能
通过正交试验筛选出的甲苯异丙基化反应条件为:原料比(甲苯/异丙醇)为4∶1,温度为200℃,空速为2h-1。在该条件下以200℃焙烧,负载量分别为30%、40% 和50% 的PWH/SiO2为催化剂,考察不同负载量对甲苯与异丙醇烷基化反应中异丙醇转化率、烷基化反应选择性的影响。
图5(a)和图5(b)分别为不同负载量PWH/SiO2催化剂催化下异丙醇的转化率和烷基化反应的选择性随时间变化的结果。由图5(a)可知,负载量对异丙醇的转化率有较大的影响。随着负载量的增加,异丙醇的转化率先增大后减小,在负载量为40% 时异丙醇的转化率最高。这是由于甲苯与异丙醇在杂多酸催化剂上的反应属于表面型反应,在低负载量下,活性中心的数目较少,催化剂活性较低,而在负载量为40% 时,磷钨酸活性组分均匀分散在载体表面,能够提供较多的活性中心,而当负载量高达50%时,磷钨酸在载体SiO2上形成多层负载,一方面造成杂多酸晶粒的聚集,质子浓度显著下降;另一方面造成部分载体孔道阻塞,反应物与催化剂接触的表面积减小,反应活性也下降。由图5(b)可知,烷基化反应的选择性随负载量的增加也呈现先升高后下降的趋势。在负载量为40% 时,烷基化 反应选择性最高,达到80% 左右。因此,对于甲苯与异丙醇烷基化反应,磷钨酸在载体SiO2上最适宜的负载量为40%,在该负载量下活性组分在载体表面既有较高的分散度,同时又保持了高密度的活性中心,因此可以发挥高效催化活性,这与XRD表征结果所获得的结论是一致的。

图5 不同负载量PWH/SiO2 对反应的影响
2.3 不同焙烧温度对PWH/SiO2 催化剂性质的 影响
浸渍法制备负载型杂多酸催化剂的过程中,焙烧温度对于杂多酸在载体表面的分散状态以及杂多酸的酸性都有重要影响。有研究报道,磷钨酸的活化温度会影响结晶水的个数,从而影响杂多酸的酸强度。在175℃处理时,磷钨酸所含结晶水为2 个,其酸强度(H0)介于-12.0~-8.2 之间;在220℃处理时,其所含结晶水为1 个, H0 介于-13.6~-12.0之间;而当处理温度高于220℃时,H0 又会逐渐降低[23]。因此本文对负载型杂多酸催化剂在不同温度下进行了焙烧处理,并对样品进行了XRD 和Py-IR表征。
2.3.1 活性组分在载体上的分散
图6 所示为负载量为40%,焙烧温度分别为160℃、200℃和260℃的PWH/SiO2的XRD 谱图。
由图6 可以看出,随着焙烧温度的升高,磷钨酸活性组分的峰强度也随之变化。当温度达到260℃时,出现较强的磷钨酸特征吸收峰。这可能是由于温度的升高使得活性组分之间的聚集程度加大,不利于其在载体表面的分散。但若焙烧温度过低,杂多酸中水含量太高,不利于催化剂酸强度的提高,因此应选择合适的焙烧温度。由图6 可知焙烧温度为200℃时活性组分在硅胶上的分散情况较好,因此确定200℃为适宜的焙烧温度。
2.3.2 催化剂的酸性
图7 为负载量为40%,焙烧温度分别为160℃、200℃和260℃的PWH/SiO2催化剂的Py-IR 谱图。从图7 中可以看出,磷钨酸PWH 具有很强的B 酸,随着焙烧温度的升高,1540cm-1处的B 酸峰强度先增大后减小,200℃焙烧温度下,B 酸和L 酸都保持了较高的酸量。而达到260℃后,B 酸峰强度反而降低,这可能是由于过高的焙烧温度使得杂多酸发生聚集而降低了表面酸性位数量。

图6 不同焙烧温度PWH/SiO2(负载量40%)催化剂的XRD 图
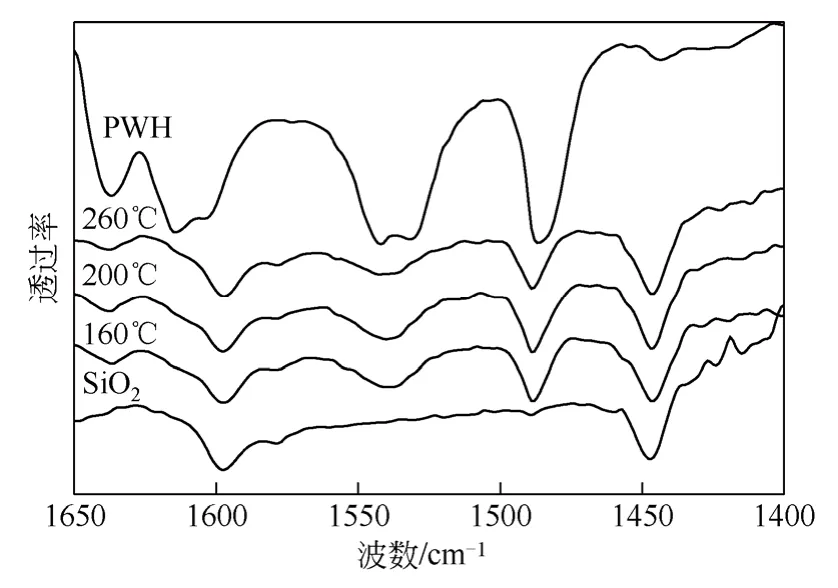
图7 不同焙烧温度PWH(40%)/SiO2 催化剂的Py-IR 图
2.3.3 不同焙烧温度催化剂的甲苯异丙基化反应 性能
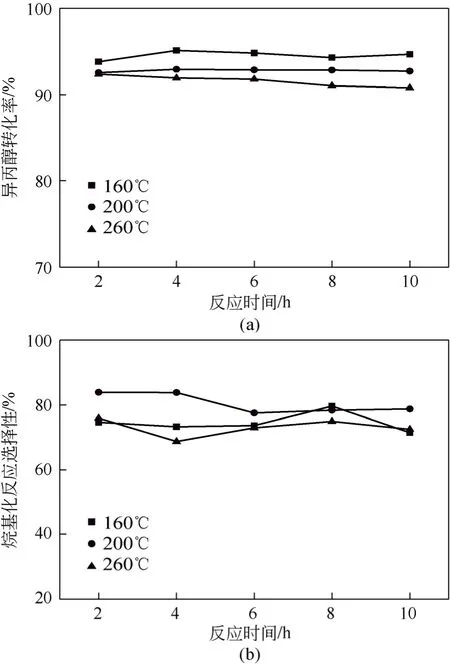
图8 不同焙烧温度PWH/SiO2 对反应的影响
图8(a)和图8(b)分别为不同焙烧温度PWH/ SiO2(负载量40%)催化下异丙醇的转化率和烷基化反应的选择性随时间变化的结果。由图8(a)可知, 焙烧温度对催化剂活性的影响较为显著,异丙醇的转化率随焙烧温度的升高而降低,160℃下焙烧所得催化剂异丙醇的转化率最高。由图8(b)可知,200℃下焙烧所得催化剂烷基化反应选择性最高。焙烧温度达到260℃时,异丙醇的转化率及烷基化反应选择性都较差。这主要是由于不同焙烧温度影响杂多酸表面结晶水的数目及其在载体SiO2表面的分散。焙烧温度为260℃,导致磷钨酸在载体表面聚集,比表面积减小,且高温下强酸的酸强度增加,而甲苯烷基化反应主要发生在弱酸和中强酸位。在200℃下,焙烧的催化剂烷基化反应的选择性最高,说明在该温度下焙烧,磷钨酸活性组分在载体表面分散情况较好,酸强度适中,因此选择200℃为负载型磷钨酸PWH/SiO2催化剂的焙烧温度。
2.4 负载型杂多酸PWH/SiO2 催化剂稳定性的初步测试
将杂多酸负载于载体上能够很大程度上实现催化剂的回收再利用,但负载型杂多酸催化剂在反应过程中仍然存在活性组分的流失问题,所以评价催化剂性能的指标还包括催化剂的稳定性。
在固定床微反装置上考察负载型杂多酸PWH/SiO2的稳定性,反应时间为24h。异丙醇的转化率及烷基化反应选择性结果如图9 所示,产物中各异构体的选择性如图10 所示。结果显示,在所考察时间内,异丙醇的转化率稳定在90%以上,烷基化反应的选择性随着反应的进行呈现逐渐下降的趋势。产物中p-IPT 的选择性随时间无明显变化,其含量维持在37%左右。随着反应的进行,产物中m-IPT 的选择性逐渐下降,o-IPT 的选择性逐渐上升,分析原因可能是随着反应的进行,催化剂表面的酸性位有所降低,酸性位的减少使得生成的o-IPT难以转化为m-IPT。装置运行24h 的结果表明,催化剂能保持较高的催化活性。
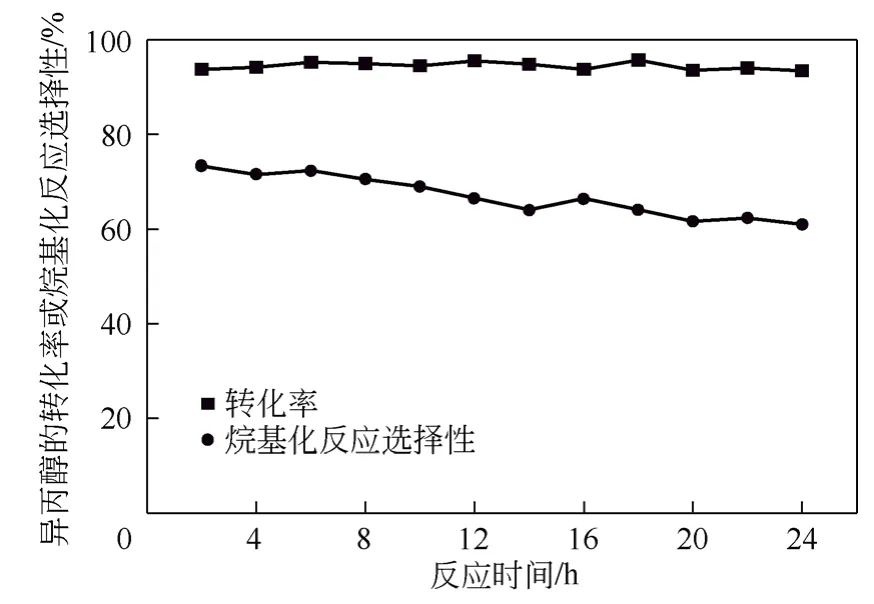
图9 异丙醇的转化率和烷基化反应选择性随时间的变化
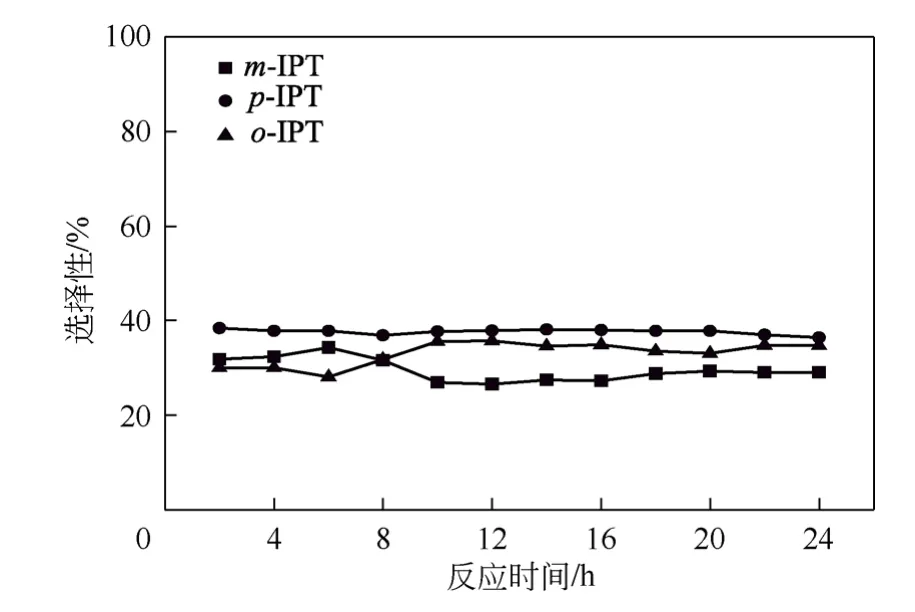
图10 异丙基甲苯各异构体随时间的变化
3 结 论
(1)负载量及焙烧温度对活性组分磷钨酸在载体SiO2上的分散及PWH/SiO2的酸强度有显著影响,通过XRD、低温N2吸附及Py-IR 表征表明,较适宜的负载量为40%,合适的焙烧温度为200℃。
(2)对不同负载量及不同焙烧温度下PWH/SiO2催化剂进行活性评价表明,负载量为40%,焙烧温度为200℃所制备的PWH/SiO2催化剂活性最高,该催化剂对异丙醇的转化率最高达92.94%,烷基化反应选择性最高达83.89%。
(3)对PWH/SiO2催化剂稳定性进行了初步测试,连续反应24h 的结果表明,催化剂仍然具有较高的催化活性,负载型催化剂催化活性稳定。
[1] 郭汝生,李书纹. 甲苯和丙烯选择烃化合成低邻位异丙基甲苯[J]. 天然气化工:C1 化学与化工,1995,20(3):39-43.
[2] 唐祥海,朱瑞芝. 甲苯-丙烯烷基化合成异丙基甲苯新型催化剂的研究[J]. 南开大学学报:自然科学版,1998,31(4):68-72.
[3] 王岩,李建伟,李英霞,等. β 分子筛上甲苯与丙烯烷基化反应的研究[J]. 化学反应工程与工艺,2010(3):248-252.
[4] 张海燕,代跃利,蔡蕾. 杂多酸催化剂催化氧化脱硫研究进展[J]. 化工进展,2013,32(4):809-815.
[5] Das J,Parida K M. Heteropoly acid intercalated Zn/AI HTlc as efficient catalyst for esterification of acetic acid using n-butanol[J]. J. Mol. Catal. A:Chem.,2007,264:248-254.
[6] Kaur J,Griffin K,Harrison B,Kozhevnikov I V. Friedel-Crafts acylation catalysed by heteropoly acids[J]. J. Catal.,2002,208:448-455.
[7] Azizi N,Saidi M R. Highly efficient ring opening reactions of epoxides with deactivated aromatic amines catalyzed by heteropoly acids in water[J]. Tetrahedron,2007,63:888-891.
[8] Dias A S,Lima S,Pillinger M,et al. Acidic cesium salts of 12-tungstophosphoric acid as catalysts for the dehydration of xylose into furfural[J]. Carbohydrate Research,2006,341:2946-2953.
[9] Hernández-Cortez J G,Manríquez Ma,Lartundo-Rojas L,et al. Study of acid-base properties of supported heteropoly acids in the reactions of secondary alcohols dehydration original research article[J]. Catalysis Today,2014,220-222(3):32-38.
[10] Rosa María Ladera,José Luis García Fierro,Manuel Ojeda,et al. TiO2-supported heteropoly acids for low-temperature synthesis of dimethyl ether from methanol Original Research Article[J]. J. Catal.,2014,312(4):195-203.
[11] Siddhartha Kumar Bhorodwaj,Dipak Kumar Dutta. Activated clay supported heteropoly acid catalysts for esterification of acetic acid with butanol[J]. Applied Clay Science,2011,53(2):347-352.
[12] Rafiee E,Paknezhad F,Shahebrahimi Sh,et al. Acid catalysis of different supported heteropoly acids for a one-pot synthesis of β-acetamido ketones original research article[J]. J. Mol. Catal. A:Chem.,2008,282(1):92-98.
[13] 李贵贤,宋维维,马建军,等. 硅烷化凹凸棒黏土负载杂多酸盐催化环己烷氧化反应[J]. 化工进展,2011,30(7):1494-1497.
[14] 欧国勇,杨辉荣,方岩雄. 负载型杂多酸催化剂研究进展[J]. 化工进展,2001,20(8):18-21.
[15] Cheng W C,Luthra N P. NMR study ofthe adsorption of phosphomolyb dates on alumina[J]. J. Catal.,1988,109(1):163-169.
[16] Rao K M,Gobetto R,Lanniello R,Zecchina A. Solid state NMR and IR studies of phosphomolybdenum and phosphomngsten heteropoly acids supported on SiO2,T-A12O3and SiO2-A12O3[J]. J. Catal.,1989,119(2):512-516.
[17] Mastikhin V M,Terkikh W,Timofeeva M N,Krivomchko O P.1H,31P NMR MAS,infrared and catalytic studies of heteropolyaeid H3PWl2O40supported on MgF2[J]. J. Mol. Catal. A,1995,95:135-140.
[18] 伊万·科热夫尼科夫. 精细化学品的催化合成:多酸化合物及其催化[M]. 北京:化学工业出版社,2005.
[19] 凌云,杜泽学,张永强. 甲苯与丙烯烷基化反应催化剂和工艺的研究[J]. 化工进展,2003,22(2):122-125.
[20] 李琴. 二氧化硅负载杂多酸催化剂的制备,表征及催化性质[D]. 青岛:中国石油大学,2008.
[21] 陈霄榕,李永丹. SiO2与Keggin 杂多酸相互作用的研究[J]. 分子催化,2002,16(1):60-64.
[22] Lefebvre F.31P MAS NMR study of H3PW12O40supported on silica:Formation of (≡SiOH2+)(H2PW12O40-)[J]. Journal of the Chemical Society:Chemical Communications,1992(10):756-757.
[23] 杜泽学. 几种杂多酸及其盐的结构和酸性特征与应用研究[D]. 北京:石油化工科学研究院,1997.
