氯过氧化物酶的研究及应用新进展
2014-07-21张丽华
张丽华
(山西大同大学化学与环境工程学院,山西 大同 037009)
氯过氧化物酶(Chloroperoxidase,CPO)是一种具有多种催化活性和广泛底物适应性的血红素过氧化物酶,目前被认为是过氧化物酶家族中应用最广泛的酶之一。它是用H2O2等过氧化物(ROOH)作电子受体,催化各类底物发生氧化反应的酶。1961年,Hager等[1-2]首次从海洋真菌(Calderiomyces fuma⁃go)培养液中分离检测出CPO。其相对分子质量约为42000 Da,等电点(pI)在 3.2 ~ 4.0 之间[3]。由于CPO独特的活性位点结构,可催化多种反应,如卤化、过氧化、氧插入反应,以及将过氧化氢分解为氧和水。另外,CPO具有高的立体选择性,能催化烯烃发生环氧化。CPO被广泛应用于手性化合物的合成、石油净化、染料降解和电化学应用等领域,对于氯过氧化物酶的研究具有重要意义。
1 CPO的结构
CPO的晶体结构如图1,不同颜色表示其次级结构的一些关键元素,紫色表示锰紧挨着中心血红素。CPO含有一条单一的多肽链,这条多肽链由299个氨基酸组成且折叠成8个螺旋状。由于远端血红素中近端配体半胱氨基酸和过氧化氢有结合点,导致CPO与细胞色素P-450的分子结构有一定相似度;但由于CPO独特的活性位点结构,使它与其他的血红素过氧化物酶和细胞色素P-450有着本质的差别,从而显示出CPO的催化多样性。CPO分子中血红素的“远端”是以谷氨酸(Glu183)而非组氨酸来起催化作用的,同时在形成化合物Cpd I的过程中,还起到断开O-O键的作用[4-5]。CPO分子中的血红素上还包含一个疏水基团,此疏水基团增强了其与底物的结合能力。
1995年,Sundaramoorthy[4]首次公开CPO的晶体结构图。研究显示,CPO活性位点的辅基是一个高自旋原卟啉IX分子。铁原子和吡咯环的4个N原子进行5配位,半胱氨基酸(Cys29)残基的硫原子代替组氨酸的咪唑基作为亚铁血红素的第5轴向配体。整个血红素是夹在C-末端和N-末端螺旋的表面间。完整的CPO结构是由8个α-螺旋结构组成的,命名为A~H。CPO的远端残基位于F螺旋中;CPO的近端位于A螺旋垂直于血红素平面且紧挨着N-末端,这是CPO区别于其他过氧化物酶和细胞色素P-450的关键点。目前,我们仍然不清楚影响CPO性质的排序机理[5]。
类似于其他过氧化物酶,血红素近端配体的稳定性由蛋白质环境所决定。尤其是CPO半胱氨基酸上硫原子的稳定性受带正电的环境影响,所以其稳定性可能和血红素中铁原子的还原电势的增加有关[6]。CPO的还原电势依赖于pH值:pH值为5.9时,其还原电势为-140 mV;pH值为2.7时,其还原电势为+150 mV。研究发现初始化合物I的形成对pH值不敏感,但化合物I的减少速率随着pH值的减少其还原电势呈增长趋势[7]。

图1 CPO的晶体结构
CPO活性位点的侧视图见图2,包括CPO中一些重要的氨基酸,如谷氨酸(Glu183)、组氨酸(His105)、半胱氨酸(Cys29)、苯丙氨酸(Phe103)和苯丙氨酸(Phe186)。然而,CPO远端不存在通常存在于其他过氧化物酶中的组氨酸和精氨酸。但很有趣的是His105距离血红素丙酸酯为3.5Å,同时His105也是锰的一个配体[8-9]。因此,His105在CPO发生催化反应时是至关重要的。Glu183和His105形成H键,两个Phe残基可与亲水底物相互作用。

图2 CPO活性位点的侧视图
跨入CPO表面表象的片段如图3所示,包括挨着蛋白质的糖类。2个箭头代表通向活性位点的2条通道,用洋红色表示血红素。卤化物的结合位点2(黄,溴;蓝,碘)位于窄通道和通道末端碘化物特异结合位点3之间,表明卤化物从表面进入血红素需通过这条窄通道。从血红素活性中心延伸到酶表面有2条底物进入通道:一条窄通道和一条宽通道,进入方式取决于其物理特性。大多数卤化物的结合位点位于窄通道,这表明了活性铁中心卤化物的进入通道。窄通道是由血红素中心庞大的疏水性残基组成,且由远离活性中心的小的极性残基组成。这样的结构便于其进入血红素活性位点,也便于特定底物取向进入血红素活性位点。尽管一个底物位点接近血红素,但血红素基团的丙酸酯边缘不利于底物的进入,所以,CPO活性中心允许底物和血红素铁中心在一个特定的位置发生立体选择性催化反应[5]。
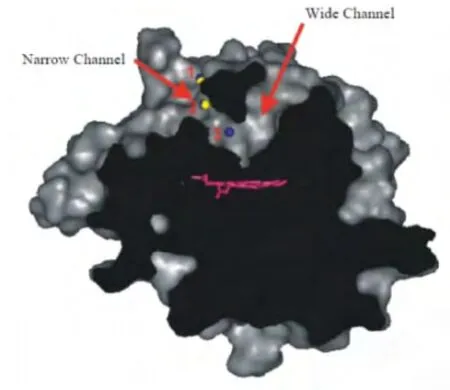
图3 通过CPO表面表象的片段
2 CPO的反应类型
在血红素过氧化物酶家族中,CPO是发生反应类型最多的一种酶。第1类是卤化,用H2O2或其他过氧化物作为引发剂,卤化物(除氟外)和易发生卤化反应的化合物作为底物(图4)[10]。第2类与第1类非常相似,除了底物是尿酸之外。这两种反应在初始步骤可能会形成一个类似氯化的中间体,因此有最佳的酸性pH值[11-12]。第3类是一个典型的过氧化反应,根据参与反应有机底物的特性决定反应的最佳pH值在4~7之间[13]。根据酶反应的特异性,氯过氧化物酶可催化H2O2发生歧化反应。最新研究表明,CPO能催化像细胞色素-P450的单加氧酶反应(图5)。另外,Dawson[14]发现CPO还能发生特定卤代芳香族化合物的催化脱卤反应。

图4 氯过氧化物酶催化卤化反应
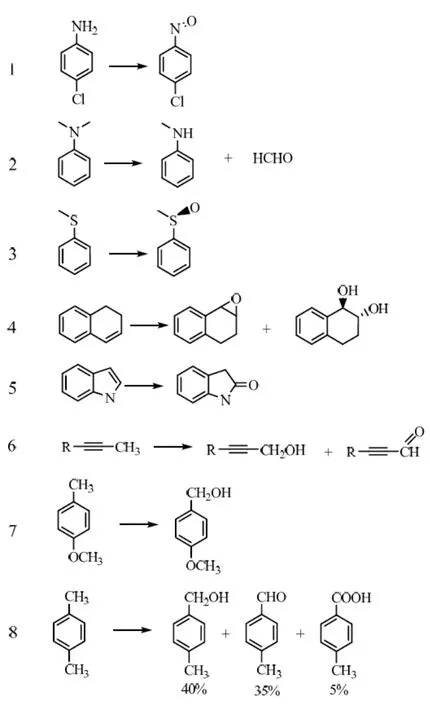
图5 氯过氧化物酶的催化氧化反应
近年来,由于CPO能合成手性药物,巨大的工业潜在价值吸引了国内外众多人士的眼球。据报道,CPO还能发生手性环氧化反应、磺化氧化反应和羟基化反应,并具有较高的产率[15-17]。
CPO发生手性催化反应的优点是不需要提供NAD(P)H或其他电子供体,但在细胞色素P-450发生催化反应时,必须提供。因此,CPO独特的特性使其在工业应用中,尤其是在手性药物的合成,石油净化和农药生产等领域具有光明的应用前景[18-20]。
3 CPO的固定化
由于CPO价格昂贵,易失活,且难以回收,导致其成本高,大大限制了其工业应用。因此,提高CPO的稳定性和重复使用率尤为重要,所以CPO的固定化成为当今酶工程领域的研究热点之一,关于CPO的固定化方法,近年来有一些报道。2010年,Jung[21]等在酸性条件下,以原硅酸四乙酯(TEOS)和非离子型嵌段共聚物P123为原料,合成大孔有序介孔分子筛SBA-15,再经修饰产生酶共价结合点。进一步将CPO和葡萄糖氧化酶固定在此介孔分子筛上,用于吲哚的氧化实验中,发现固定化CPO存储数周不失活。此外,在固定床反应器中连续操作,发现固定化酶不易浸出,这解决了工业应用中酶的回收问题。2009年,Zhang[22]等通过共价交联法,以壳聚糖为载体,戊二醛作交联剂,制备了壳聚糖膜(球)固定化CPO。实验表明,固定化酶热稳定性、对化学变性剂尿素和过氧化氢的耐受性均明显提高。
磁性高分子微球是近年研制的一种新型高分子磁性固定化材料,是通过共聚或表面改性等方法将磁性无机粒子与有机高分子结合形成的具有一定磁响应性及特殊结构的复合微球。Zhang等[23]通过共价结合法以xMagTM异硫氰酸根末端磁性微球为载体固定CPO,pH为5.5,10 min后,酶载量可达25.59 mg/g,固定化率为75.5%。相对天然 CPO 而言,固定化CPO米氏常数Km值增大,与底物亲和力减小,热稳定性、抗尿素、H2O2及各种有机溶剂的失活能力明显增强,而且固定化CPO在重复使用16次后,其活力还有85%以上,远远高于天然CPO。
4 氯过氧化物酶的应用
4.1 染料废水
印染与染料废水成分复杂,具有COD高,色泽深、酸碱性强、水质变化大等特点,其净化一直是当今世界难题。随着染料工业的迅猛发展,我国的水环境也受到严重的污染。那么寻求经济,绿色环保的净化方法已迫在眉睫,研究表明采用生物酶催化降解是极具发展潜力的绿色方法。2012年,张娟等[24]研究了H2O2存在的情况下,CPO对偶氮染料(橙黄Ⅳ和皂黄)的催化氧化。结果表明,加入H2O2,CPO对橙黄Ⅳ和皂黄具有较好的脱色降解效果,橙黄Ⅳ在5 min的脱色率达91.02%,皂黄在7 min的脱色率达88.83%。
4.2 手性化合物的合成
手性化合物是一类特殊的化合物,具有独特的性质,通常含手性因素的化学药物所具有的对映异构体在生物体内的药理活动,代谢过程及毒性存在显著差异,如一个是药物,而另一个却有毒。由于CPO对有机底物具有手性识别功能,所以,CPO在手性药物制备和新型手性催化剂的开发等领域具有潜在的价值。2012年,汪丽敏等[25]用CPO作催化剂,甲基苄基硫醚为原料,叔丁基过氧化氢(TBHP)为氧化剂,直接一步合成手性R-苄基甲基亚砜的反应。结果表明:少量添加剂的引入可有效改善CPO的催化性能。
4.3 石油净化
石油在提炼净化过程中大致产生4类产品,分别为石油燃料,润滑油和润滑脂,沥青质和石油焦,石油溶剂和石油化工原料。沥青质是原油中最顽固的部分,利用化学法处理污染严重,使用生物酶处理,绿色环保且效果明显。1993年,Fedorak等[26]首次用生物酶CPO对富含石油卟啉环的沥青质进行酶法处理。2012年,Ayala[27]等在水含量为6.3%的三元体系中,利用CPO催化原油中的沥青质馏分。实验表明,用壳聚糖共价交联CPO后,可使本体系的总转换数(TTN)提高10倍。荧光光度计显示,用CPO处理过的沥青质中的芳香基降低了24%,表明生物转化后的显著变化。另一方面,溶解度谱分析表明:相对于未处理的沥青质而言,经CPO处理的沥青质更难溶于甲苯中,在己烷存在下更容易产生沉淀物,这可能与极性原子的引入有关。能量色散型X射线光谱仪(EDS)表明:氯含量在原子基础上增加了6倍。最后,在氮气气氛下的热降解证明,经CPO处理的沥青质更易反应,从而产生更少的焦炭。
4.4 电化学领域
CPO固定到金、玻碳和热解石墨电极表面,表现出良好的直接电子传递作用。CPO在催化底物的同时,氧分子被还原成水,在生物电子设备的研制中成为理想的生物电极阴极催化剂,如在神经发育、血压控制方面。所以,CPO在电化学领域具有良好的应用前景。2012年,田海涛等[28]将CPO与双十二烷基溴化铵(DDAB)混合,涂于Nation修饰的玻碳电极表面,于表面再涂上壳聚糖,最后制得Chi/CPO-DDA B/Nafion/GC修饰电极。研究显示,CPO与电极之间产生了直接的电子传递。说明CPO能催化氧化肉桂醇,生成肉桂醛。2010年,穆世磊等[29]利用双十二烷基二甲基溴化铵将CPO固定于经Nafion预处理的玻碳电极上。研究表明CPO可进行准可逆的电子传递反应,能将甲苯催化氧化为苯甲醛。
5 存在的问题及展望
作为一种生物催化剂,CPO具有良好的应用前景。它在工业应用中备受瞩目,但各国学者对其研究还远远不够,国内关于CPO的报道更是鲜见。CPO的结构和催化机理、如何通过物理和化学等手段进一步提高CPO的利用率还有待研究。今后可以考虑从以下几方面进行探索:第一,CPO的结构以及发生不同反应的催化机理,应通过先进的分析方法和手段对反应过程中的中间体和活性自由基进行分析测定,详述反应机理;第二,探讨CPO的来源、结构和物化性能的关系,为其工业化生产打下理论基础;第三,尝试更多的载体固定CPO,减少酶失活同时能最大程度保持酶活力。更深入地研究CPO基础理论及尽快使已取得的实验成果上升到应用阶段,将成为今后研究CPO的重点。
[1]Fang G H,Kenigsberg P,Axley M J,et al.Cloning and sequencing of chloroperoxidase cDNA[J].Ncleic Acids Research,1986,14:8061-8071.
[2]Shaw P D,Hager L P.Biological Chlorination.VI Chloroperoxidase:A component of the β-Ketoadipate chlorinase system[J].J Biol Chem,1961,236:1626-1630.
[3]Pickard M A,Hashimoto A.Stability and carbohydrate composition of chloroperoxidase from caldariomyces fumago grown in a fructose-salts medium[J].Can J Microbiol.1988,34:998-1002.
[4]Sundaramoorthy M,Terner J,Poulos T L.The crystal structure of chloroperoxidase:a heme peroxidase-cytochrome P450 funtional hybrid[J].Structure,1995,3:1367-1377.
[5]Kuhnel K,Blankenfeldt W,Terner J,et al.Crystal Structures of Chloroperoxidase with Its Bound Substrates and Complexed with Formate,Acetate,and Nitrate[J].J Biol Chem,2006,281:23990-23998.
[6]Adachi S,Nagano S,Ishimori K,et al.Roels of Proximal Ligand in Heme-Proteins-Replacement of Proximal Histidine of Human Myoglobin with Cysteine and Tyrosine by Site-Directed Mutagenesis as Models for P-450,Chloroperoxidase,and Catalase[J].Biochemistry,1993,32:241-252.
[7]Makino C R,Hager L P.Oxidation-reduction potential measurements on chloroperoxidase and its complexes[J].Biochemistry,1976,15(21):4748-4754.
[8]Mroczko Y X,Manoj M,Wang K M,et al.Replacement of the proximal heme thiolate ligand in chloroperoxidase with a histidine residue[J].Proc Natl Acad Sci USA,1996,96:12412-12417.
[9]Conesa Y X,Punt A,Hager L P.Examining the Role of Glutamic Acid 183 in Chloroperoxidase Catalysis[J].J Biol Chem,2003,278:13855-13859.
[10]Hallenberg P F,Hager L P.Purification of chloroperoxidase from Caldariomyces fumago[J].Meth Enzymol,1978,52:521-529.
[11]Shahangian S,Hager L P.The reaction of chloroperoxidase with chlorite and chlorine dioxide[J].J Biol Chem,1981,256(12):6034-6040.
[12]Libby R D,Thomas J A,Kaiser L W,et al.Chloroperoxidase halogenation reactions.Chemical versus enzymic halogenating intermediates[J].J Biol Chem,1982,257(9):5030-5037.
[13]Sun W,Kadima T A,Pickard M A,et al.Catalase activity of chloroperoxidase and its interaction with peroxidase activity[J].Biochem Cell Biol,1994,72(7):321-331.
[14]Dawson J.Chloroperoxidase.Evidence for P-450 type heme environment from magnetic circular dichroism spectroscopy[J].J Am Chem Soc,1976,98:3709-3710.
[15]Allain E J,Hager L P,Deng L,et al.Highly enantioselective epoxidation of disubstituted alkenes with hydrogen peroxide catalyzed by chloroperoxidase[J].J Am Chem Soc,1993,115:4415-4416.
[16]Dexter A F,Lakner F J,Campbell R A,et al.Highly Enantioselective Epoxidation of 1,1-Disubstituted Alkenes Catalyzed by Chloroperoxidase[J].J Am Chem Soc,1995,117:6412-6413.
[17]Zaks A,Dodds D R.Chloroperoxidase-catalyzed asymmetric oxidations:substrate specificity and mechanistic study[J].J Am Chem Soc,1995,117:10419-10424.
[18]Dexter A F,Lakner F J,Campbell R A,et al.Highly Enantioselective Epoxidation of 1,1-Disubstituted Alkenes Catalyzed by Chloroperoxidase[J].J Am Chem Soc,1995,117:6412-6413.
[19]Miller V P,Tschirret-Guth R A,Montellano P R.Chloroperoxidase catalyzed benzylic hydroxylation[J].Arch Biochem Biophys,1995,319(2):333-340.
[20]Hager L P,Lakner F J,Basavapathruni A.Chiral synthons via chloroperoxidase catalysis[J].J Mol Cat B:Enzymatic,1998,5:95-101.
[21]Jung D,Streb C,Hartmann M.Covalent Anchoring of Chloroperoxidase and Glucose Oxidase on the Mesoporous Molecular Sieve SBA-15[J].Int J Mol Sci,2010,11:762-778.
[22]Zhang L H,Bai C H,Wang Y S,et al.Improvement of chloroperoxidase stability by covalent immobilization on chitosan membranes[J].Biotechnol Lett,2009,31:1269-1272.
[23]张丽华.氯过氧化物酶的固定化及其应用研究[D].西安:陕西师范大学,2008.
[24]张娟,蒋育澄.氯过氧化物酶催化过氧化氢氧化水溶性偶氮染料的降解[J].工业催化,2012,20(2):70-75.
[25]汪丽敏,吴金跃,蒋育澄,等.手性苄基甲基亚砜的氯过氧化物酶催化定向合成[J].化学学报,2012,70(4):465-470.
[26]Fedorak P M,Semple K M,Vazquez-Duhalt R,et al.Chloroperoxidasemediated modifications of petroporphyrins and asphaltenes[J].Enzyme Microb Technol,1993,15:429-437.
[27]Ayala M,Hernandez-Lopez E L,Perezgasga L,et al.Reduced coke formation and aromaticity due to chloroperoxidase-catalyzed transformation of asphaltenes from Maya crude oil[J].Fuel,2012,92:245-249.
[28]田海涛,王珂,李琳,等.氯过氧化物酶修饰电极催化氧化肉桂醇的研究[J].化学研究与应用,2012,24(11):1636-1641.
[29]穆世磊,田海涛,陆中庆.氯过氧化物酶修饰电极对甲苯的催化氧化[J].化工学报,2010,61(s1):20-23.
