金属-石墨烯复合物吸附活化CO2的第一性原理
2014-07-18陈小康齐晶瑶
李 欣,陈小康,齐晶瑶
(1.哈尔滨工业大学理学院,150001哈尔滨;2.哈尔滨工业大学市政环境工程学院,150090哈尔滨)
金属-石墨烯复合物吸附活化CO2的第一性原理
李 欣1,陈小康1,齐晶瑶2
(1.哈尔滨工业大学理学院,150001哈尔滨;2.哈尔滨工业大学市政环境工程学院,150090哈尔滨)
在光催化CO2的反应体系中,CO2的吸附活化是一个关键步骤;不同的活化方式和CO2的活化态决定了其反应路线和最终产物.以金属-石墨烯体系为研究对象,采用密度泛函理论方法,结合局域密度近似(LDA)和PWC泛函,计算该体系在CO2吸附前后的几何结构、能量、电荷分布和态密度等的变化.结果表明:电子从金属-石墨烯体系转移到CO2,使CO2带负电并活化;其中Cu-G体系对CO2的活化效果最好,C—O键长分别增加6和14 pm,O—C—O键角减小为122°;金属原子簇和石墨烯的第一电离能和电子亲和势对电子的转移起决定性作用,金属原子簇电子亲和势比石墨烯第一电离能越大,电荷越易从石墨烯转移到金属原子簇.
二氧化碳;石墨烯;金属;催化剂;第一性原理计算
目前,由于化石燃料的广泛使用,大气中二氧化碳浓度逐年迅速增加,已成为一个严重的全球问题[1].另一方面,由于煤炭、石油和天然气等的大量消耗也引起了全球范围内的能源短缺问题.因此,对CO2进行有效回收和利用具有解决能源及环保问题的双重意义[2].迄今为止,在众多的转换方法中,通过光催化反应将CO2转化为具有高附加值的化工产品或有机燃料是最有发展前途的有效技术手段[3].具有碳最高氧化态的CO2分子十分稳定,它的活化和转化需要相当苛刻的反应条件.在光催化CO2体系中,催化剂载体除了应具有稳定性等特点外,还应具有很强的吸附能力,使CO2在催化剂表面尽可能地被吸附和活化,以提高光催化反应的量子效率.石墨烯因具有稳定的结构、独特的光电特性、较大的比表面积和极强的吸附能力已成为理想的光催化剂载体[4].
近年来,量子化学理论研究的迅速发展以及计算机性能的快速提高,使得较精确地处理复杂的催化体系成为可能.本文在第一性原理计算的基础上,研究金属-石墨烯体系对CO2的吸附和活化过程,通过理论计算和分析进行催化剂的初步筛选.
1 计算方法
所有的密度泛函理论(DFT)计算均通过运行DMol3代码[5-6]实现.交换相关函数采用局域密度近似(LDA)[7-9]和PWC[10]泛函,计算金属纳米粒子、石墨烯以及金属-石墨烯复合体系的电子结构、几何结构和能量.计算时选用DND基组,并结合有效势芯(ECP)[11-12]近似处理方法.为了确保计算结果具有可比性,孤立的CO2分子、原始的石墨烯、金属与石墨烯的复合物和吸附CO2的金属石墨烯体系均采用相同的计算条件.由于研究的所有体系主要是沿着与石墨烯片层平行的方向伸展,为了提高计算精度,所有体系k点分布设置为2×2×1,以增加在xy平面上的k点数目.计算中采用导体近似屏蔽模型(COSMO)[13-15]作为溶剂化模型,并将水(相对介电常数εr=78.54)作为溶剂,模拟溶剂对所有体系的影响,使结果更加符合真实的环境.设定最大收敛能量差为5.442×10-4eV(能量1 Ha=27.21 eV),最大位移为0.4 pm,最大的力为13.605 eV·pm-1,用于确定自洽场迭代是否已经收敛.
在计算模拟过程中,为了更准确地模拟实际体系,均使用三维周期性边界条件.6×6(铜石墨烯复合物和金石墨烯复合物体系)或7×7(银石墨烯复合物体系)单层石墨烯超胞结构分别用于研究体系,同时在两层石墨烯层之间建立2 nm的真空层.这些设定可以削弱相邻超胞中石墨烯层和金属原子簇之间的相互作用,以排除它们之间相互作用对结果的影响.除了在对CO2-M-G(CO2吸附在金属-石墨烯复合物上的体系)体系进行结构优化时限制了M-G(金属-石墨烯复合物体系)原子的坐标,其他所有优化和能量计算过程中,原子的所有坐标均完全释放.为了讨论金属原子簇与石墨烯之间的相互作用强度,定义了金属原子簇和石墨烯相互作用能Ei,即

式中:EM-G为金属原子簇与石墨烯结合后体系的整体能量;EM为金属原子簇的能量;EG为石墨烯的能量.
3种金属的晶体结构均为面心立方(fcc).所有的金属原子簇由9个金属原子构成,与石墨烯接触的面为金属的(100)面.4个金属原子在簇合物的顶层,5个在底层,这与类似结构相关实验保持一致[16-17].从理论上说,这种碟状几何结构可以增强金属原子簇和石墨烯之间的相互作用,也可以尽量降低M-G体系的表面能.
2 M-graphene体系
2.1 几何结构
所有金属簇M和金属簇石墨烯复合物(MG,M是Cu、Ag和Au)均采用相同的方法进行优化,优化结果如图1所示.可以看出,金属原子簇几何结构相对原始金属结构只发生了很小的形变,由5个金属原子组成的底层发生了扭曲.中心原子向下突出,与底层中的其他4个原子组成了类似金字塔的结构.Cu、Ag和Au金属簇合物的中心原子与底层周围4个金属原子形成的平面之间的距离dz分别为13.8,21.1和34.0 pm.这可能是由于没有周围金属原子的作用,这些金属原子受力不平衡.如表1所示,为了降低结构中的张力,金属原子簇的几何形状向凸多面体的结构发生形变,缩短了所有M-M的键长,特别是位于表面上键长明显缩短也证实了这一猜想.这种变形也可以降低原子簇的表面积,通过缩短所有键的长度,使体系结构向球形方向发生形变,有效地降低体系的能量.在俯视图中可以看出,顶层和底层周围4个原子的几何结构都接近正方形.
在M-G复合物中,Cu和Ag原子簇结构变化很大,对称性由C4v变成了C2v.Cu、Ag金属原子簇中底层中心原子与周围原子形成平面之间的距离也大幅度减小,分别由13.8 pm和21.7 pm变为2.6 pm和22.4 pm.底层原子几何结构接近平面结构,这可能是因为石墨烯与金属原子簇之间的相互作用平衡了底层原子的受力.而Au-G体系中Au原子簇结构几乎没有变化,这与Au同石墨烯之间很弱的相互作用相一致.
在M-G复合物体系中,石墨烯中的C原子相对于石墨烯C原子的平均位置均出现一定的偏离.在Cu-G、Ag-G和Au-G体系中,相对的平均位置最大偏差分别为20.2,14.4和13.2 pm,即石墨烯形变量的大小关系为Cu>Ag>Au.在一定程度上,C原子偏离平衡位置的距离,揭示了金属原子簇和石墨烯之间相互作用的强度.这种形变可能是因为底层金属原子与石墨烯中C原子之间发生轨道重叠,使得靠近金属簇的C原子的杂化类型发生部分转变,即从sp2杂化部分转变为sp3杂化.

图1 金属原子簇和金属-石墨烯体系优化后的几何结构

表1 金属簇合物体系几何结构
总之,金属团簇和石墨烯之间的相互作用越强,石墨烯的形变就越明显,更有利于平衡底层金属原子的受力情况,金属原子簇中的底层将更加平坦.
2.2 结合能
考察所有体系的总能量.表2给出了石墨烯与不同金属原子簇的结合能.可以看出,所有金属原子簇与石墨烯的结合过程从能量角度讲是自发的,Cu-G、Ag-G和Au-G体系的结合过程能量变化分别为-2.75,-3.34和-1.98 eV(1 eV= 96.49 kJ/mol).即使考虑与石墨烯相互作用的底层原子数目为5,它们之间的相互作用依然很强.此外,金属簇与石墨烯之间的相互作用越强,说明体系更稳定,更易于合成.
从表2还可以看出,石墨烯与金属原子簇之间的结合能与电荷量的绝对值之间存在正相关关系,说明两者复合时,电荷的转移与两者的相互作用之间存在一定的内在联系.
表2 M-G体系中金属原子簇电荷量和金属原子簇与石墨烯之间的结合能
2.3 Mulliken电荷分布与电子转移
Mulliken布局分析结果表明,金属原子簇底层中心原子带正电,铜、银和金团簇分别约为0.024,0.028和0.119 e,底层的其他4个原子带负电,而顶层的4个原子带正电.因此,底层周围4个原子附近的电子密度高于底层中心原子和顶层4个原子附近的电子密度.其中Au原子簇的电子密度分布差异最大.在一定程度上,金属原子簇不同位置的原子附近电子密度的微小差异是吸附和催化性能明显不同的根源.当金属簇和石墨烯结合之后,Cu-G和Ag-G体系中金属簇底层中心原子带更多的正电荷(分别增加0.037和0.109 e),底层其他4个原子电性发生了反转,且Ag-G体系中的金属簇电荷变化更大.此时,顶层的4个原子具有不同的电荷性质,两个体系中更接近中心的两个原子比其他两个原子具有更多的电子.从总体上看,铜、银原子簇与石墨烯结合后,分别有0.217和0.687 e的电子从金属簇转移到石墨烯.而Au-G体系的电荷分布和转移情况与Cu-G和Ag-G体系完全不同,当Au金属簇与石墨烯结合后,底层中心原子带有更少的正电荷,底层其他原子所带负电荷的量减少,而顶层原子电荷发生了翻转.整体电子不是有金属转移到石墨烯,而是石墨烯向Au原子转移电子0.190 e.
M-G体系中,Ag原子簇的电荷分布类似于Cu原子簇.但是,Ag原子簇顶层原子电荷分布的不均匀性大大增加,换句话说,所有原子具有更多的剩余电荷.另外,在Ag-G体系中,从金属簇到石墨烯的电荷转移量大于Cu-G体系.由于CO2是一个强电子受体,这种电荷转移将不利于吸附在Ag-G复合物上CO2的活化,Ag原子簇中低电子密度的d轨道不能有效地将电子转移到CO2的π∗轨道上.Au-G体系中,Au原子簇上具有更多的电子,电子密度越大,越利于金属原子的d轨道与CO2的π∗轨道的重叠.但是Au原子簇带有更多的负电荷,从另一个方面也说明其具有更大的电子亲和势,不利于CO2从Au金属原子簇上获得电子.
复合物中石墨烯的电荷分布变化也很大.原始的石墨烯碳原子是中性的.然而,与金属原子簇结合后,金属原子簇与石墨烯之间发生了电子转移,使得石墨烯带上电荷,并且越靠近金属原子簇的C原子带有更多的电荷.此外,从图2中也可以看出,Cu-G体系中的石墨烯电荷分布最不均匀.与金属原子直接接触的C原子带负电荷,而间接接触的原子却带上了正电荷,远离金属原子的C原子接近中性.这体现了金属原子簇与石墨烯之间电荷转移的细节,也体现出Mulliken布局分析的缺陷.Mulliken布局分析的缺陷在Au-G体系中石墨烯电荷分布中体现得非常明显.电荷是从石墨烯转移到金属,与金属原子直接接触的原子失去电子,应该带正电荷.但是由于Mulliken布局分析是将两个原子共有额外电荷均分给两个原子,这样必然会将金属原子弥散的电子云中的电子不合理地分配给C原子,造成这些C原子带上负电荷.Ag-G体系中石墨烯的电荷分布与Cu-G体系类似,只是不均匀性减小.而Ag-G体系中电荷转移量最大.由此说明,电荷分布的不均匀性与电荷转移量之间没有必然联系,应该是与之间的相互作用和金属原子性质有关.
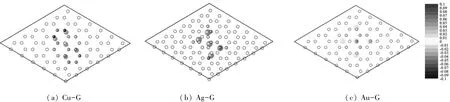
图2 M-G体系中石墨烯的Mulliken电荷分布
如表3所示,石墨烯和金属原子簇的第一电离能、电子亲和势和Fermi能级绝对值的整体趋势为:Au>G>Cu>Ag.电子将尽可能填充到能量较低的能级上,因此,Au-G体系中,电子必然从石墨烯转移到Au原子簇.而在Cu-G和Ag-G体系中,电子从金属原子簇转移到石墨烯,从而使金属簇整体带正电.

表3 石墨烯与金属原子簇第一电离能、电子亲和势、费米能级和金属功函实验值eV
2.4 态密度
M-G体系各部分和整体的PDOS(偏态密度)如图3~5所示.从石墨烯(6×6)的PDOS(图3(b))中可以看出,随着能量从-19 eV增加到-3 eV,s轨道的DOS(态密度)减为0,而p轨道的DOS一直增加.能量大于-3 eV的态密度几乎完全归功于C原子p轨道的贡献.在约-3 eV的位置存在一个分割区,能量低于-3 eV的DOS对应石墨烯中C原子的sp2杂化轨道,-3 eV以上对应于C原子2pz轨道的DOS.在费米能级以上,孤立的尖峰对应石墨烯的π∗轨道.

图3 Cu-G体系各部分和整体的PDOS
比较石墨烯和Cu-G的DOS发现,当Cu原子簇与石墨烯结合后,-6 eV附近宽阔的p带整体向低能量区域发生了移动,即更多的p轨道(pz轨道)与s轨道杂化.这与几何结构分析结果相符,即石墨烯中一些sp2杂化的C原子部分地向sp3杂化转变.π∗带向低能量方向迁移,说明在Cu-G体系中,更多的电子填充在石墨烯的π∗带,降低了Cu-G体系中石墨烯的稳定性.总体来说,Cu原子簇和石墨烯的结合将一些sp2杂化的C原子部分转变为sp3杂化,部分打破了π电子的离域,因此,石墨烯的稳定性大大降低.此外,Cu金属簇与石墨烯结合后,Cu原子的d带略微变宽,并与pz带强烈共振,说明Cu金属簇中Cu原子的d轨道和石墨烯的pz轨道间存在强烈相互作用.Cu原子簇d带尖峰的消失也证明了它们之间存在强烈的相互作用.

图4 Ag-G体系各部分和整体的PDOS
Cu和Ag在周期表中属于同一族,具有相似的价电子结构,Ag原子簇与石墨烯之间也存在强烈的相互作用,这种作用同样源于Ag原子簇d带和石墨烯C原子pz带的共振.Ag-G体系中,石墨烯的DOS向低能量区域移动更加明显,换句话说,Fermi能级移动到能量更高位置,即更多的石墨烯DOS填充了电子.这可能是由于Ag原子簇的DOS更加分散,更多的轨道分布在费米能级之上.费米能级以下轨道数量太少,不能容纳Ag原子簇的所有电子,因此,电子填充到具有更高能量的轨道.同时,费米能级附近的DOS强度非常弱,增强了这种迁移.与Cu原子簇的d带相比,在Ag -G复合物中Ag原子簇的d带变化很小,小于Cu -G复合物.说明Ag-G体系中的金属簇d带与石墨烯p带之间的相互作用比Cu-G体系弱,这与从几何结构分析得到的结果相符.
Au原子簇合物DOS中,-7~0 eV的宽带主要为d带.0 eV以上为Au原子簇的6s带.Au原子簇的d带变窄,尖峰突出,体现了电荷转移对能带结构的影响.同样,Au原子簇d带与石墨烯pz带强烈共振.说明Au原子簇d轨道与石墨烯C原子的pz轨道之间发生重叠.

图5 Au-G体系各部分和整体的PDOS
3 CO2-M-G体系
3.1 几何结构
为了研究M-G体系对CO2分子的吸附活化效果,对CO2-M-G(CO2吸附在M-G体系上的体系,M为Cu、Ag和Au)体系进行了结构优化和性质计算.
CO2吸附在M-G上优化后的几何结构如图6所示.在CO2-Cu-G体系中,CO2以桥位吸附在Cu原子簇上,CO2中的一个C和一个O原子分别与Cu原子簇中的一个Cu原子相连.
C—Cu和O—Cu键长均约为193 pm,Cu—O和Cu—C共价半径之和分别为175和187 pm.然而,在CO2-Ag-G体系中,CO2和Ag原子簇之间的键长分别为245和258 pm,均明显大于Ag—C共价半径之和203 pm.上述分析表明,CO2和Cu原子簇之间形成了化学键,而CO2和Ag原子簇之间很可能没有键合作用.
如表4所示,在CO2-M-G体系中,本来直线型的CO2分子发生了弯曲,Cu、Ag和Au体系中键角∠OCO分别为122°,144°和170°.同时,相对于正常CO2键长(实验值为114 pm,相同条件下的计算值为117 pm),Cu、Ag体系中CO2的两个C—O键长明显增大.吸附在Cu-G体系上CO2的两个C—O键长分别为123和131 pm,在CO2-Ag-G体系中均为121 pm.这些形变说明,吸附在M -G体系上的CO2分子与金属原子簇之间存在强烈的相互作用.而Au体系中CO2的键长几乎没有变化,为119 pm.说明Au-G体系与CO2的相互作用很弱,活化作用也很弱.
由于CO2分子结构的改变,CO2分子中C和O原子的p轨道不能有效重叠,sp杂化的碳原子部分转变为sp2杂化,进而削弱了C=O之间的双键.由此可以推出,M-G体系活化了CO2分子,使其更易于受其他物质的进攻,进而能发生化学反应.比较吸附在Cu-G、Ag-G和Au-G上CO2的键长和角度可以得出一个结论,Cu-G的活化效果最好.

图6 CO2-M-G体系优化后的几何结构
3.2 电荷分布与电子转移
CO2吸附在M-G上后,对体系中各金属原子上面的电荷分布进行分析.当CO2吸附在Cu-G和Ag-G体系上后,CO2带较多的负电荷.Cu-G和Ag-G体系中CO2上的多余电荷分别为-0.733和-0.446 e.因为CO2分子是一个强电子受体,使CO2带上负电荷能够破坏其原始结构,使其活化,最终催化其与其他物质发生化学反应.

表4 CO2-M-G体系各部分电荷分布和CO2分子几何结构
CO2分子活化的根本原因为:多余的负电荷意味着较多的电子,这些电子可以填充在CO2的2πu反键轨道上,削弱CO2分子的C=O键,进而CO2分子可以与其他分子碰撞或自身振动发生化学反应.
3.3 态密度
CO2-M-G的PDOS如图7~9所示.可以看出,CO2的能带与金属原子簇的d带存在强烈的共振,这从另一个方面说明CO2分子与金属原子簇之间存在强烈的相互作用.此外,与金属d带共振的主要是CO2的p带,说明金属原子主要同CO2的p轨道发生作用.由于金属d轨道的作用,不同程度地破坏CO2分子中p轨道形成的π键,此为CO2在金属原子簇上吸附活化的根源.
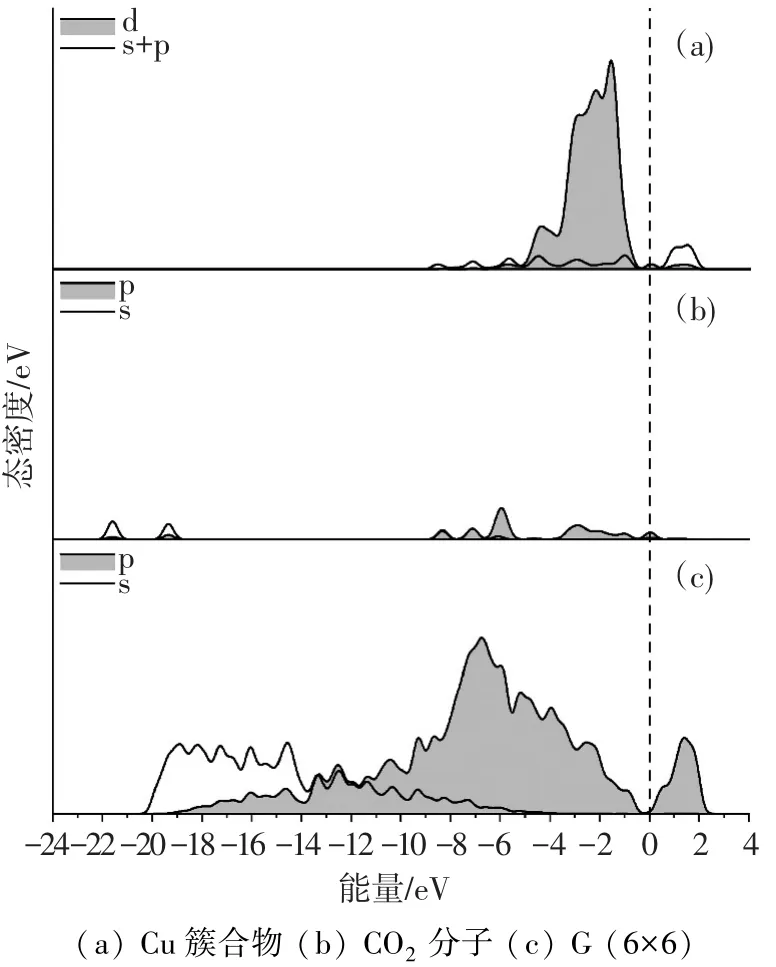
图7 CO2-Cu-G体系各部分的PDOS
比较Cu、Ag和Au体系的DOS图可以看出,CO2分子的p带与Cu-G体系中金属簇d带的共振最强烈.说明Cu-G体系对CO2π键的破坏程度最大,因而CO2的活化效果最好.

图8 CO2-Ag-G体系各部分的PDOS
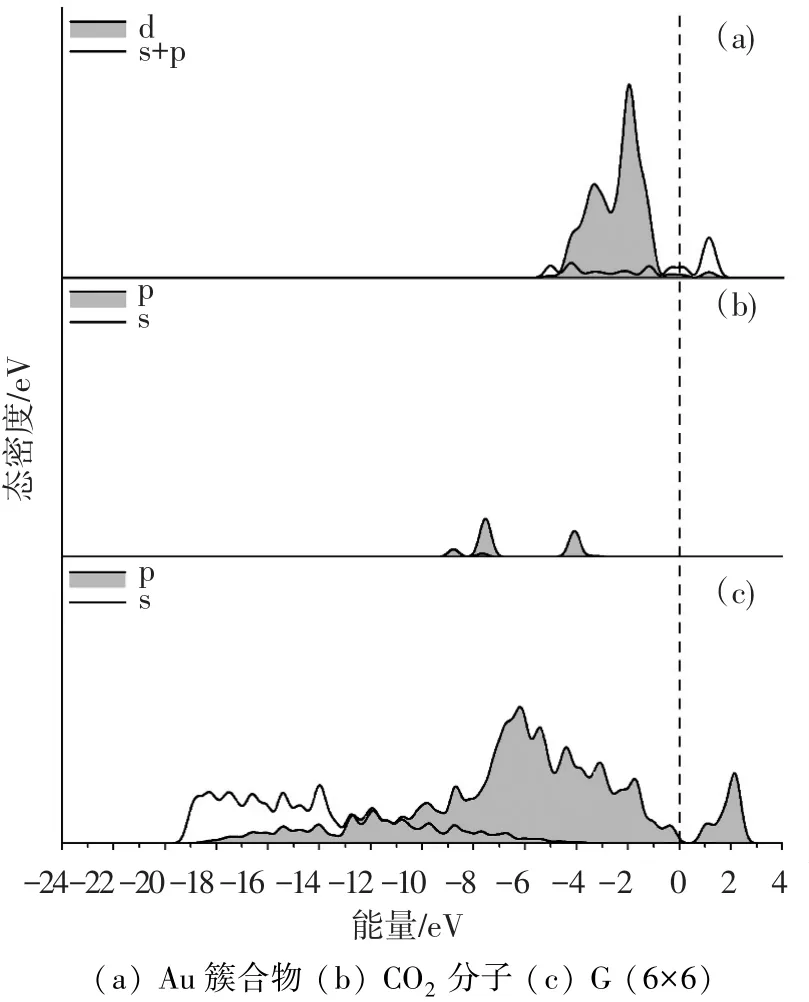
图9 CO2-Au-G体系各部分的PDOS
4 结 论
1)金属原子簇与石墨烯的结合能均很大.同时,金属原子簇与石墨烯之间的结合能与两者之间电荷转移量正相关.
2)Cu、Ag和Au簇合物与石墨烯形成的3种复合物中,Cu-G体系对CO2活化效果最好.CO2活化的根源为金属原子簇的d带与CO2的p带相互作用,最终破坏了CO2分子中p轨道形成的π键.
3)M-G体系对CO2的活化性能与最终转移到CO2上的电子直接相关,主要取决于金属原子簇的第一电离能和电子亲和势.金属原子簇必须具有适当的电子接受能力才能使金属石墨烯体系达到最好的CO2活化效果.
[1]LAL R.Sequestration of atmospheric CO2in global carbon pool[J].Energy Environ Sci,2008,1:86-100.
[2]王明明,徐磊,段雪,等.中国二氧化碳资源化有效利用的战略选择[J].资源科学,2009,31(5):829-835.
[3]倪小明,谭猗生,韩怡卓.二氧化碳催化转化的研究进展[J].石油化工,2005,34(6):505-512.
[4]LIUHaitao,RYUS,CHENZheyuan,etal. Photochemical reactivity of graphene[J].J Am Chem Soc,2009,131:17099-17101.
[5]DELLEY B.An all-electron numerical-method for solving the local density functional for polyatomic-molecules[J].J Chem Phys,1990,92:508.
[6]DELLEY B.From molecules to solids with the DMol3approach[J].J Chem Phys,2000,113:7756.
[7]HEDIN L,LUNDQVIST B I.Explicit local exchange correlation potentials[J].J Phys C,1971,4:2064-2083.
[8]CEPERLEY D M,ALDER B J.Ground state of the electron gas by a stochastic method[J].Phys Rev Lett,1980,45:566-569.
[9]LUNDQVIST S,MARCH N E.Theory of the inhomogeneous electron gas[M].Plenum:New York,1983.
[10]PERDEW J P,WANG Yue.Accurate and simple analytic representation of the electron-gas correlation energy[J].Phys Rev B,1992,45:13244.
[11]DOLG M,WEDIG U,STOLL H,et al.Energyadjusted abinitiopseudopotentialsforthe1st-row transition-elements[J].J Chem Phys,1987,86:866.
[12]BERGNER A,DOLG M,KUECHLE W,et al.Abinitio energy-adjusted pseudopotentials for elements of groups 13-17[J].Mol Phys,1993,80:1431.
[13]SLATER J C.Statistical exchange-correlation in the selfconsistent field[J].Adv Quantum Chem,1972,6:1-92.
[14]KLAMT A,SCHÜÜRMANN G.COSMO:a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient[J]. J Chem Soc,Perkin Trans,1993,2:799.
[15]DELLEY B.The conductor-like screening model for polymers and surfaces[J].Mol Simul,2006,32:117-123.
[16]NURIA L,JENS K N.Catalytic CO oxidation by a gold nanoparticle:a density functional study[J].J Am Chem Soc,2002,124:11262-11263.
[17]HÖLZL J,SCHULTE F K.Work function of metals,in solid surface physics[M].Springer-Verlag:Berlin,1979.
(编辑 刘 彤)
First-principle theory calculations of CO2adsorption and activation by metal-graphene composite
LI Xin1,CHEN Xiaokang1,QI Jingyao2
(1.School of Science,Harbin Institute of Technology,150001 Harbin,China;2.School of Municipal and Environmental Engineering,Harbin Institute of Technology,150090 Harbin,China)
Metal-graphene system was taken as the research object.Density functional theory(DFT),combined with local density approximation(LDA)and PWC functional,was employed to study the changes in the geometry structure,energy,charge distribution and density of states(DOS)of the systems before and after absorption of CO2on them.The results show that the electrons are transferred from the M-graphene system to CO2,which is eventually activated by negative charge.The Cu-G system is most effective to activate CO2in these three complexes.The bond length of CO2increases by 6 and 14 pm,respectively,and the bond angle of O—C—O decreases to 122°.Furthermore,the first ionization energy and electron affinity of metal clusters and graphene play a decisive role in the electron transfer.Compared with the first ionization energy of graphene,the larger the electron affinity of metal clusters,the more the electrons transferred from graphene to metal cluster.
carbon dioxide;graphene;metal;catalyst;first-principle theory calculation
O641.12
A
0367-6234(2014)10-0058-07
2013-06-20.
国家自然科学基金资助项目(51178142).
李 欣(1965—),男,教授,博士生导师;齐晶瑶(1960—),女,教授,博士生导师.
李 欣,lixin@hit.edu.cn.
