外电场作用下L1OH 分子的特性研究
2014-07-13吴学科梁冬梅
吴学科,梁冬梅,2,荆 涛
(1.凯里学院物理与电子工程学院,凯里556011;2.山东大学化学与化工学院,济南250100)
1 引 言
L1OH,又称为3,5 二甲氧基苯甲醇(3,5-dimethoxybenzyl alcohol),是一族树枝状分子LnOH(整数n表示“树枝”的层数)的起始物,也是一种医药中间体.已有不少文章[1-5]报道了L1OH的结构和性质,但从目前的文献来看,对L1OH在外场作用下的激发态特性研究还未见报道,掌握L1OH 分子在一定外场作用下的激发规律,对于深入研究同L1OH 相关的药物及树枝状分子具有重要的意义[6].L1OH 分子在没有外电场情况下的几何机构如图1所示,本工作根据文献[5]理论计算出的振动频率值和实验观测值较好吻合的结果,采用与文献[5]相同的密度泛函理论B3LYP方法在6-311G(d,p)基组水平上,对L1OH 分子在X 轴方向上依次加入了强度为0.01、0.02、0.025、0.03和0.04a.u.的外电场进行几何结构优化,然后在X 轴方向上采用了杂化CIS-DFT 方法[7],研究了同等强度的外场下L1OH 分子的激发能,激发波长和振子强度.其中1 a.u.=5.14225×1011V/m.
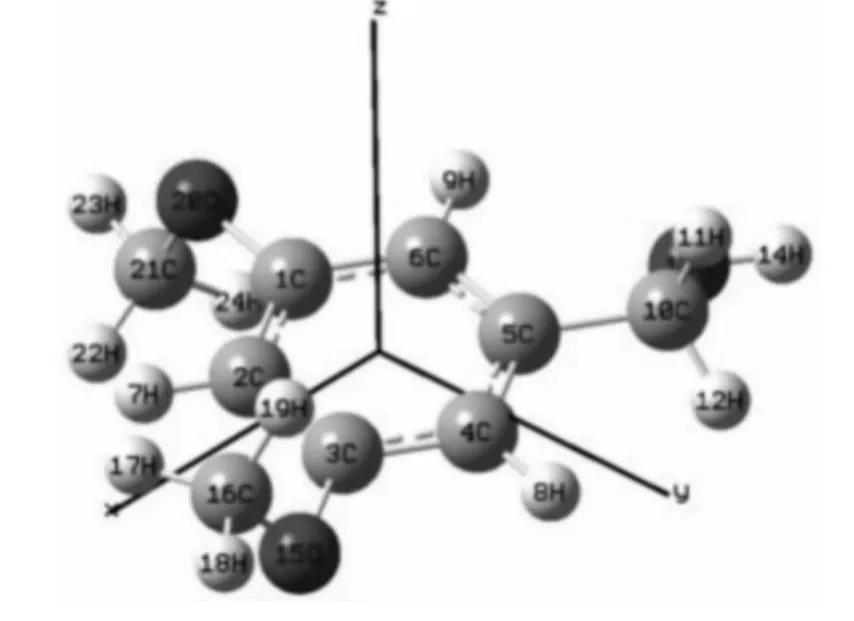
图1 无外电场作用下L1OH 分子的结构Fig.1 Optimized ground state geometry for L1OH without electric field
2 理论和计算方法
根据分子光谱学,微扰作用时间内的含时薛定谔方程可以写成[8]
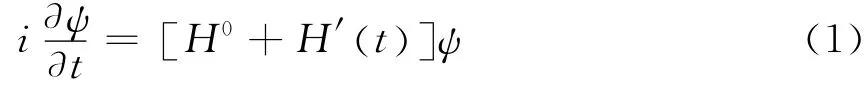
其中 H′(t) 为体系与辐射相互作用对原哈密顿量的附加项,也就是微扰项,而分子电偶极矩可以导出辐射场与体系的相互作用项

μ 为电偶极矩.
Grozema等人提出了以下的模型[9,10],
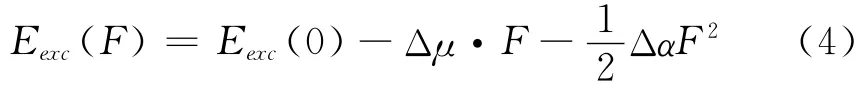
Eexc(0)是在没有电场下的激发能,其振子强度flu为[11]
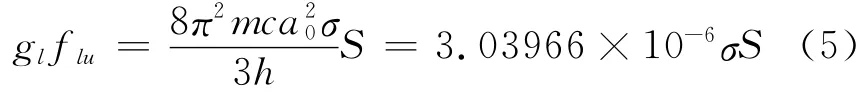
而且
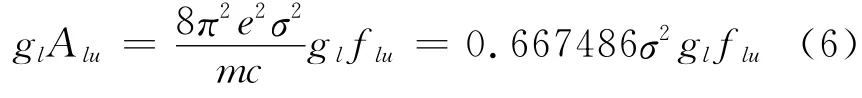
本文根据L1OH 分子的标准坐标,沿X 轴方向加强度依次为0、0.01、0.02、0.025、0.03和0.04a.u.的外电场,对其分子结构进行优化,并在优化后的基础上采用CIS/6-311G(d,p)方法对L1OH分子的激发能、波长及振子强度等进行了计算.
3 计算结果与讨论
3.1 无外电场下L1OH 分子的激发特性
首先,采用密度泛函方法中的B3LYP/6-311G(d,p)优化了L1OH 分子的基态几何结构,然后采用杂化CIS-B3PLY/6-311G 方法计算了L1OH 分子从基态跃迁到前9 个激发态的激发能,激发波长和跃迁轨道等激发参量进行了计算,结果如表1所示.
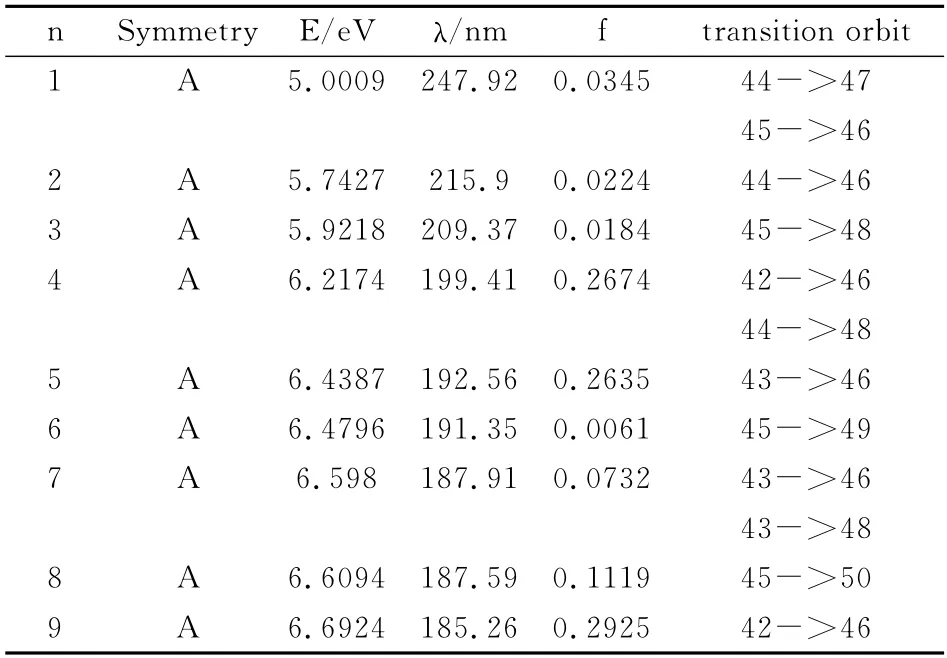
表1 L1OH 分子的激发能,激发波长,振子强度和跃迁轨道Table 1 The excitation energies,wavelengths,oscillator strengths and transition MOs of L1OH molecular
从表1 可以看出,L1OH 分子从基态跃迁到前9个激发态的激发能逐渐增大,从基态跃迁到第一激发态的激发能最小,说明其最容易激发,振子强度为0.0345,而其它振子强度都不为零,说明都可以发生跃迁,但是跃迁强弱不同,激发波长不断减小,且电子跃迁光谱集中在紫外区.
3.2 外电场对L1OH 分子的总能量、偶极矩和前线轨道能量的影响
沿如图1所示的分子坐标的X 方向加电场分别为0.01、0.02、0.025、0.03和0.04a.u.,采用密度泛函方法B3LYP/6-311G(d,p)方法对不同电场下L1OH 分子的基态构型进行了优化,得到了其总能量,偶极矩和从轨道HOMO-2,HOMO-1,HOMO,LUMO,LUMO+1到LUMO+2轨道能随外场的变化,如图2、3和4及表2所示.
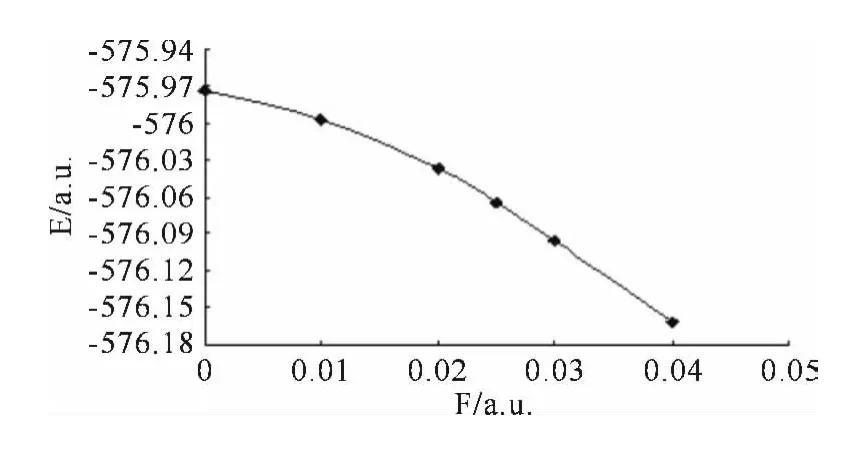
图2 分子总能量E 随电场强度变化的关系Fig.2 The energy vs electric field strength
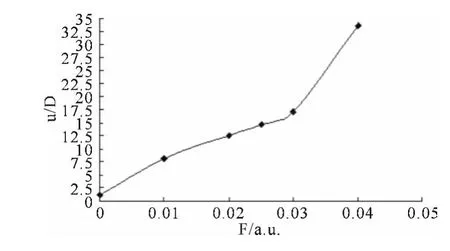
图3 分子电偶极矩μ 随电场强度变化的关系Fig.3 The dipole moment vs electric field strength
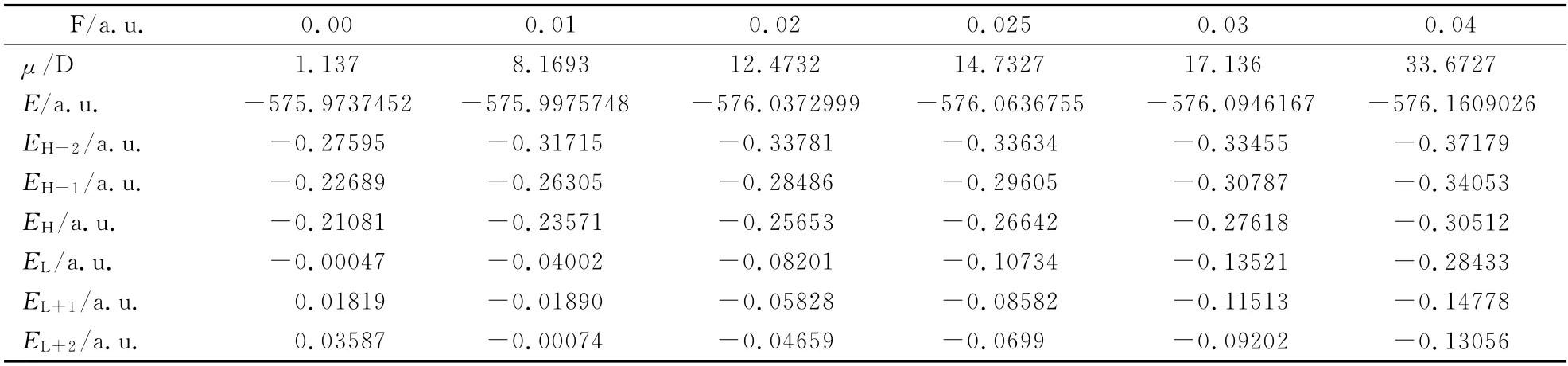
表2 L1OH 分子的偶极矩,总能量和轨道能量随外电场的变化Table 2 Optimized ground state dipole momentμ,energy Eand energy of frontier MOs of L1OH under electric fields
从图2和图3 及表2 中可以看出:L1OH 分子的总能量随外电场的不断增加而降低,从最初的-575.9737452a.u.降到-576.1609026a.u..另外,其偶极矩随外电场的增加不断增大,变化相当明显,从起初的1.1370D 增加到了最后的33.6727D,约是前者的30倍,说明其极性随外场的增加而急剧增大.
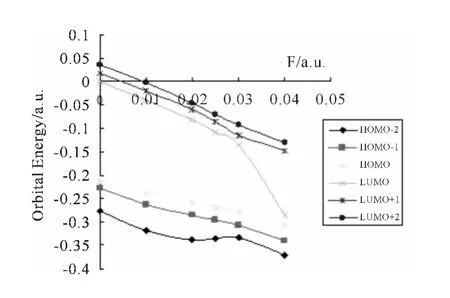
图4 不同轨道的能量随X 方向外电场F的变化Fig.4 Variation energies of MOs L1OH in response to the fields applied to the molecular
从图4中可以看出,各轨道能基本上都随外电场的增加逐渐减小.根据量子化学前线轨道理论可知,最低空轨道LUMO 轨道的能量越低,说明该分子接受电子的能力越强,而最高占据轨道HOMO 轨道能量越低,说明其轨道中的电子越稳定,越不易失去电子,能隙的大小反映了电子从占据轨道向空轨道发生跃迁的能力,从图4中可以看出虽然所有轨道的能量随外电场的增加都有所降低,但是HOMO 轨道的能量降低幅度较小,而LUMO 轨道的能量降低幅度很大,在外场为0.04a.u.的时候,LUMO 轨道的能量降低到了-0.28433a.u.,这个时候HOMO 轨道能量为-0.30512a.u.,同时也说明能隙逐渐变小,在外场为0.04a.u.的时候时L1OH 分子的能隙最小.
图5展示了最高占据轨道HOMO 和最低空轨道LUMO随外场的变化情况,在外电场不是很大的时候,从F=0.00a.u.~0.03a.u.内,HOMO轨道形状基本上保持不变,也就是说这时外场对前线轨道的分布影响不是很多,但随着外电场的继续增大,L1OH 分子的轨道分布明显受外电场的影响,从图5的0.04a.u.中可以明显看出.LUMO 前线轨道受外场的变化明显,特别是F=0.04a.u.时,轨道形状完全发生了变化,如图5所示.

图5 最高占据轨道HOMO 和最低空轨道LUMO 随外场的变化Fig.5 Evolution of frontier MOs of L1OH as a function of electric field applied to the molecular
3.3 外电场对L1OH 分子激发态的影响
在上述优化的基础上,采用杂化CIS/6-311G(d,p)方法计算了外电场下L1OH 分子从基态跃迁到前9个激发态的激发特性.(包括激发能,激发波长和振子强度)如图6和表3所示.
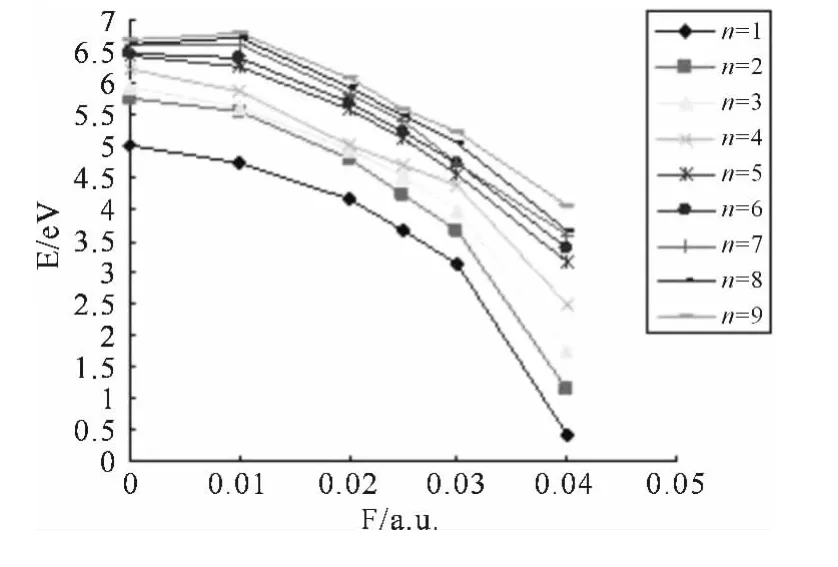
图6 L1OH 分子的激发能随外电场F的变化Fig.6 The excitation energies changing of L1OH molecular along with the field
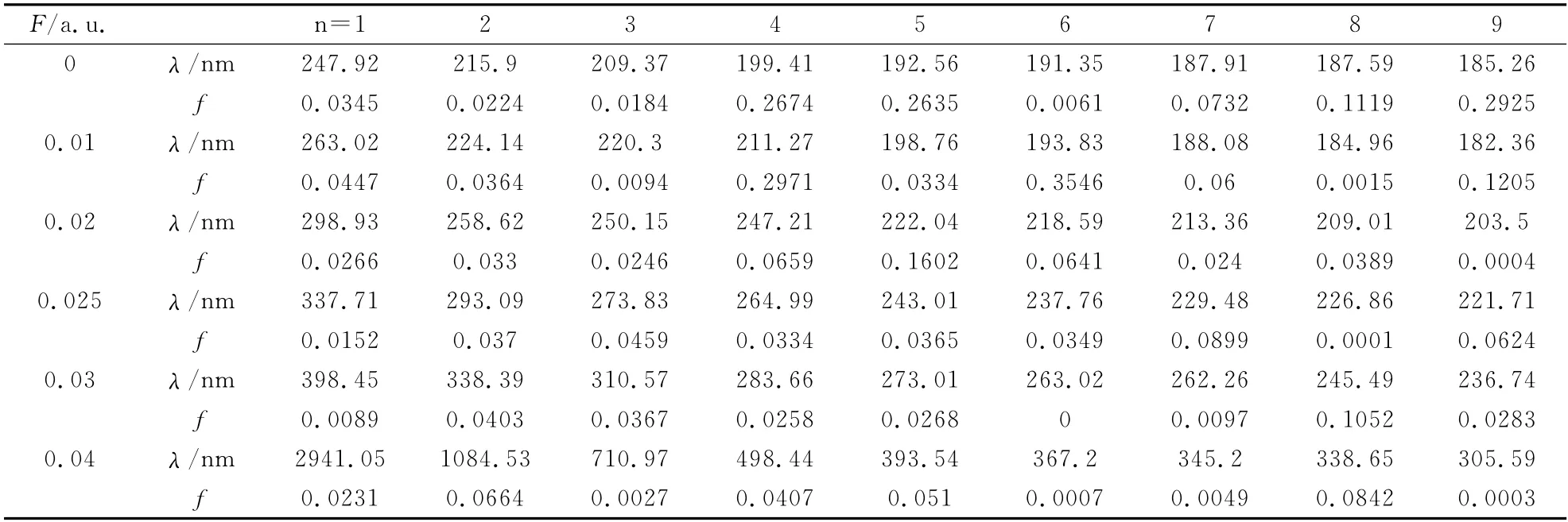
表3 L1OH 分子的激发波长与振子强度随外场F的变化Table 3 The excitation wavelengths and oscillator strengths of L1OH molecular along with the fields
从图6中可以看出,各个激发态的激发能随外电场的增加而逐渐减少,说明随外电场的增加,L1OH 分子越来越容易被激发,从表3可以看出,在F=0.00a.u.时,振子强度均不为零,都可以发生跃迁,加上不同强度的外电场后,振子强度也发生了不同的变化,如F=0.03a.u.时,第6激发态的振子强度为零,属于禁阻跃迁,在F=0.02a.u.时的第9激发态,F=0.025a.u时的第8激发态,F=0.04a.u.时,对应第9激发态的振子强度接近于零,就算是激发,也比较微弱,在实验上也可能观察不到.分析激发波长可以看出,随着外电场的不断增加,除了第8,9激发态外,其它激发态的激发波长逐渐增长,特别在F=0.04 a.u.强电场下时,第1、2激发态的激发光谱由紫外到红外的跃变,第3、4激发态的激发光属于可见光中的红光和紫光,此时的跃迁为价电子跃迁,在F<0.01a.u.时,第4~9激发态的激发光谱属于远紫外光,其它激发光谱属于紫外光.
4 结 论
本文采用密度泛函B3LYP 方法和CIS 方法对不同外电场下L1OH 分子的激发与光谱性质进行了研究,结果表明外电场对L1OH 分子的能级分布和激发特性都有一定的影响,结论如下:
(1)在没有外电场的情况下,L1OH 分子的各激发态都能够激发,激发能随级数增加而不断增大,说明越来越不容易被激发;
(2)在有外电场的作用下,L1OH 分子的总能量和偶极矩受外场的影响比较大,总能量随外电场的增加逐渐减少,偶极矩随外电场的增加不断增加,其前线轨道的能量随外电场的增加不断减少,轨道分布也受外电场很大的影响;
(3)外电场对L1OH 分子的激发也有很大的影响,如F=0.03a.u.时,第6激发态的振子强度为零,属于禁阻跃迁,在F=0.02a.u.,0.025 a.u.,0.04a.u.时,对应的第9,第8,第9激发态的振子强度接近于零,就算是激发,也比较微弱,在实验上也可能观察不到.另外,外电场对L1OH 分子的激发波长也产生了很大的影响.
[1] Pan Z,Cheung E Y,Harris K D M,et al.Structural aspects of a dendrimer precursor determined directly from powder X-ray diffraction data[J].Cryst.Growth.Des.,2004,4:451.
[2] Wong P S H,Srinivasan N,Kasthurikrishnan N,et al.On-line monitoring of the photolysis of benzyl acetate and 3,5-dimethoxybenzyl acetate by membrane introduction mass spectrometry[J].J.Org.Chem.,1996,61:6627.
[3] Gonzalez T,Terrion M C,Zapico E J,et al.Use of multiplex revers transcription-PCT to study the expression of a laccase gene family in a basidiomycet-ous fungus[J].Appl.Environ.Microbiol.,2003,69:7083.
[4] Cozens F L,Pincock A L,Pincock J A.et al.The role of nonaromatic isomers in the Photochemistry of 3,5-dimethoxybenzyl Acetate[J].J.Org.Chem.,1998,63:434.
[5] Han Y X,Shi L F,Han L G,et al.Study on the vibrational spectra of 3,5-dimethoxybenzyl alcohol[J].Spectrosc Spect Anal,2010,30:1802(in Chinese)[韩运侠,师凌枫,韩礼刚,等.3,5-二甲氧基苯甲醇振动光谱的研究[J].光谱学与光谱分析,2010,30:1802]
[6] Mo Y J,Jiang D L,Uyemura M,et al.Energy funneling of IR photons captured by dendritic antennae and acceptor mode specificity:anti-stokes resonance Raman studies on iron(Ⅲ)porphyrin complexes with a poly(aryl ether)dendrimer framework[J].J.Am.Chem.Soc.,2005,127:10020.
[7] Grimme S,Density functional calculations with configuration interaction for the excited states of molecules[J].Chem.Phys.Lett.,1996,259:128.
[8] Xu G L,Lu W J,Liu Y F,et al.Effect of external electric field on the optical excitation of silicon dioxide[J].Acta Phy.Sin.,2009,58:3058(in Chinese)[徐国亮,吕文静,刘玉芳,等.外电场作用下二氧化硅分子的光激发特性研究[J].物理学报,2009,58:3058]
[9] Grozema F C,Telesca R,Joukman H T.Excited state polarizabilities of conjugated molecules calculated using time dependent density functional theory[J].Chem.Phys.,2001,115:10014.
[10] Kjellberg P,Zhi H,Pullerits T.Bacteriochlorophyll in electric field[J].J.Phys.Chem.B,2003,107:13737.
[11] Zhu Z H,Fu Y B,GAO T,et al.Hydrogen molecule under electromagnetic and electric field[J].J.At.Mol.Phys.,2003,20:169(in Chinese)[朱正和,付依备,高涛,等.H2的外场效应[J].原子与分子物理学报,2003,20:169]
