NpFn分子结构与电子结构的理论研究
2014-07-13杜磊强武芳线董晨钟王永成
杜磊强,武芳线,董晨钟,王永成
(1.西北师范大学物理与电子工程学院,甘肃省原子分子物理与功能材料重点实验室,兰州730070;2.西北师范大学与中科院近代物理研究所极端环境原子分子物理实验室,兰州730070;3.西北师范大学化学化工学院,兰州730070)
1 引 言
随着核能的发展,核废料的处理已成为当务之急.核废料中含有的锕系元素如Np,Am 和Cm等,对环境及人体都会产生很大的危害,对这些长寿命的锕系元素的妥善处理与处置将是一个关系到今后我国核能能否大规模发展的紧要问题[1].而且锕系元素5f 电子的离域效应[2]、5f 电子对成键的贡献[3],使得锕系元素及其化合物呈现出许多不寻常的物理和化学性质.因此,获得锕系元素及其化合物完整的电子结构特性和光谱数据对于进一步探索核废料中锕系元素的处理具有非常重要的意义.
镎的氟化物作为主要锕系化合物之一,一方面由于锕系化合物的不稳定性使实验研究难度较大[4];另一方面,镎原子5f,6p,6d 和7s轨道形成的能级结构复杂,电子关联效应及相对论效应显著[5],使得对其化合物的理论研究较为困难.到目前为止,人们仅对镎氟化物分子的热力学特性和结构特性进行过一些有限的理论与实验的研究.例如Kaltsoyannis等[6]用非相对论和相对论的分离变量Xa方法研究了八面体NpF6分子基态的电子结构,同时分析了分子轨道的成分;Kimura等[7]利用电子衍射方法测量了NpF6分子的点群对称性和结构参数;Hay等[8]用78个中心电子有效芯势结合B3LYP密度泛函方法计算了NpF6分子的键长、键角和振动频率;Brown 等[9]用NpF6和碘反应制备了NpF5分子,并测出了它的晶体参数和红外光谱;Eller等[10]在NpO2与O2F2反应产生NpF6的过程中观测了中间产物NpF5分子的Raman光谱;Moritz 等[11,12]利用基于有效芯势的MOLPRO 程序包优化了NpF5和NpF3分子的结构,并进行了轨道布局分析;Ellis等[13]用全相对论自洽场方法计算了NpF4分子结构,发现了Np的5f、6d、7s和7p 轨道与F的2p 轨道间的很强的共价效应;Konings等[14]由蒸汽压测量的热力学量推得了NpF4分子的结构,同时给出了它的结构参数和光谱数据.但是,所有的这些研究获取的信息并不完整,也不成体系,且已有的实验数据与理论预测之间也存在着较大差异.因此,系统地获得镎氟化物完整的结构特性和光谱数据就显得尤为必要.
本文利用杂化密度泛函(B3LYP)方法,对F原子采用6-31+G*全电子基组,对Np原子采用具有两种不同芯电子相对论有效原子芯势(RECP)近似下的价电子基组,系统优化了NpF6、NpF5、NpF4和NpF3分子可能的几何构型,确定了对应稳定结构的能量、平衡结构参数、振动频率和红外光谱数据,并对其进行了自然键价轨道(NBO)分析.同时,比较了两种不同芯电子RECP近似下的计算结果.
2 计算方法
由于锕系元素核外电子数多,用全电子方法计算分子结构相对困难,必须采取近似方法.考虑到分子的化学性质主要由价电子决定,芯电子仅起到屏蔽核电荷和给价电子提供有效势场的作用[15],可以将价电子和芯电子的贡献分开处理.只需直接处理价电子问题的相对论有效芯势方法被认为是对锕系元素进行计算的有效方法之一.为了比较芯电子的多少对计算结果的影响,本文对Np原子采用具有两种不同芯电子RECP:一种是1998年Hay等[8]在计算NpF6的分子结构和振动光谱特性时使用的78个芯电子RECP.在这个势中,芯中包含78个电子,其余的外层电子6s、7s、6p、6d、5f 均被当做价电子处理,其收缩基函数为(7s6p2d4f)/[3s3p2d2f];另一种是德国斯图加特大学提出的60个芯电子RECP[16].这种60个芯电子的RECP代替了从n=1到n=4内壳层的60个电子,把n=5壳层(5s,5p,5d,5f),以及6s、6p、6d 和7s轨道的电子当做价电子处理,其收缩基函数为(12s11p10d8f)/[8s7p6d4f].对F原子,由于其中包含的电子的个数仅为9个,则使用了6-31+G*全电子基函数,即6-31G 加入双ζ极化函数和弥散函数.在进一步的分子结构计算中,我们利用了梯度修正的Becke交换泛函和三参数Lee,Yang和Parr 相关泛函联系在一起的B3LYP[17,18]方法.所有计算使用Gaussian03程序完成[19].
3 结果与讨论
3.1 NpFn分子的几何结构
图1给出了通过优化各个分子在不同构型下的结构而得到的分子稳定构型.其中,图1(a)~(d)给出关于NpF6、NpF5、NpF4和NpF3的结构,它们与前人[7,11,12,14]的优化结果完全一致.表1进一步给出了分别在78个和60个芯电子RECP下优化分子结构得到的结构参数.
由表1 可见,对于具有Oh点群对称性的NpF6分子,当多重度为2 时,NpF6分子能量最低,分子结构最为稳定.NBO 分析表明,未配对的一个分子轨道由Np的5f电子形成.与实验值比较发现,在60个芯电子RECP下优化的Np-F
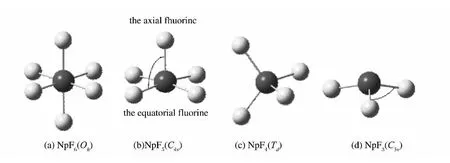
图1 NpFn分子的平衡结构Fig.1 Equilibrium structures for NpFn molecule
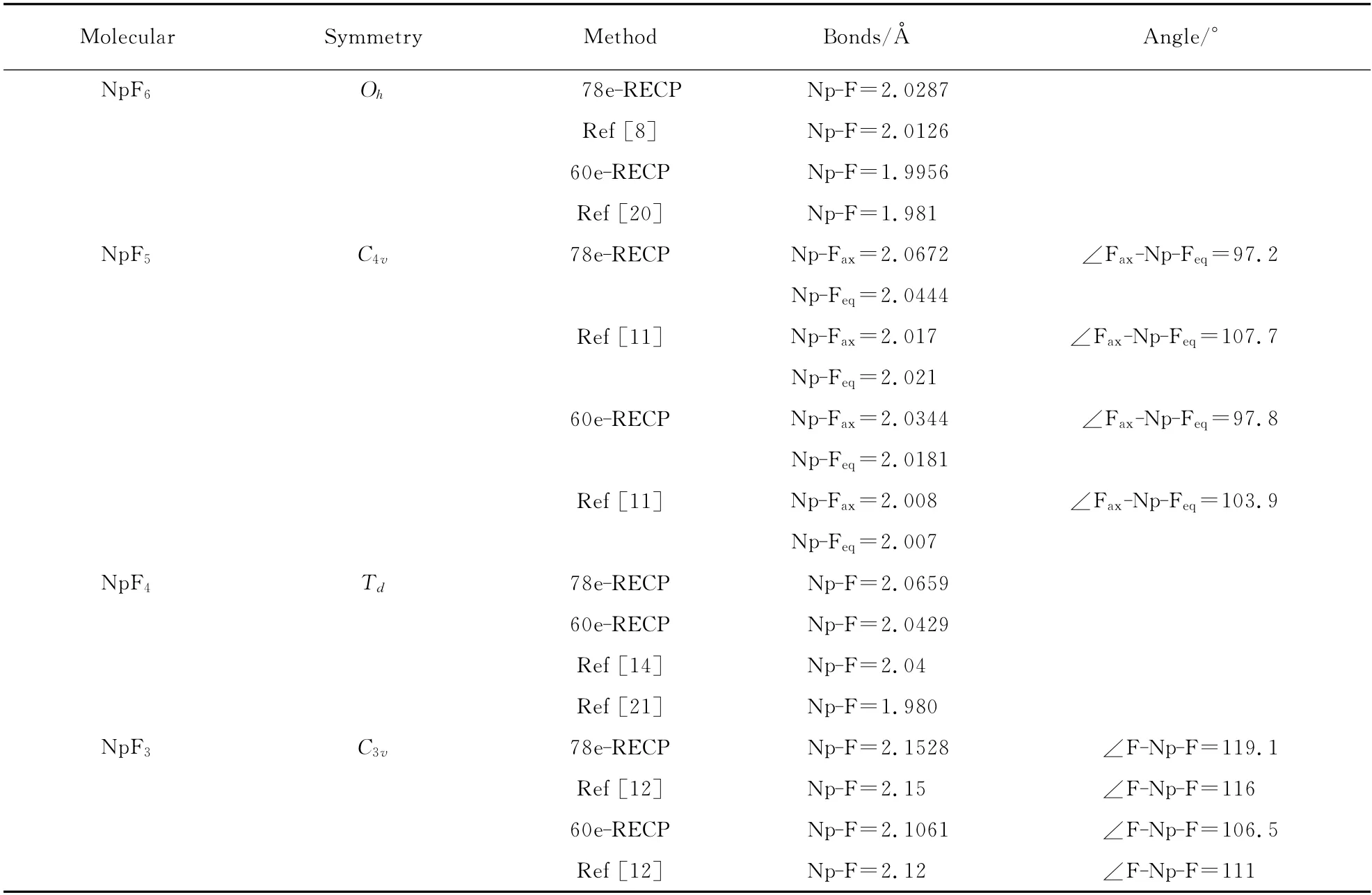
表1 NpFn分子的平衡结构参数Table 1 The equilibrium structures parameters for NpFn molecule
键长与实验值符合得更好;对于NpF5分子,在对称性为C4v、多重度为3时,具有最稳定结构.NBO 分析表明,未配对的两个分子轨道由Np的两个5f电子占据;对于具有Td对称性的NpF4分子,当多重度为4 时,能量最低.NpF4分子中未配对的三个分子轨道均由Np原子的5f电子所占据.同样,60 个芯电子RECP 下计算的Np-F 键长更接近实验值;对于具有C3v对称性的NpF3分子,多重度为5时具有最稳定的结构.NpF3分子中未配对的四个分子轨道由Np的5f电子占据.比较发现,60个芯电子RECP 下计算的分子键长比78个芯电子RECP下计算的却要短些,且更接近实验值,同时各种分子的Np-F 键长随着F 原子数的减少依次增长.
表2给出了NpFn分子振动频率和红外(IR)强度的计算结果,并与可行的理论值和实验值进行了比较.可以看出,除NpF5分子的υ2(A1)和NpF3分子的υ2(A1)及υ4(E)振动模式的计算值与实验结果和参考文献值相差较大外,其它分子的计算值与参考文献符合较好,且在60 个芯电子RECP下,多数频率的计算值比在78 个芯电子RECP下的计算结果大了约10%,个别的甚至达到50%以上,但分子键长的值却都变短了约2%.此外,从表1和表2可以看出,60个芯电子RECP下的结果比78个芯电子RECP下的结果与实验数据符合得更好.NpF5分子的υ2(A1)振动模式下的频率值与已有的两个实验值的差别较大,可能与两个已有的实验结果之间本身差别较大有关.NpF3分子的υ2(A1)和υ4(E)振动模式的计算值与参考文献相差较大,原因可能是由于文献中给出的频率值是通过与该元素分子量相似的元素的相应振动频率的实验值建立的方程式外推所得到的,可能有较大的误差.当然,目前计算结果的精确性还有待进一步的理论和实验验证.
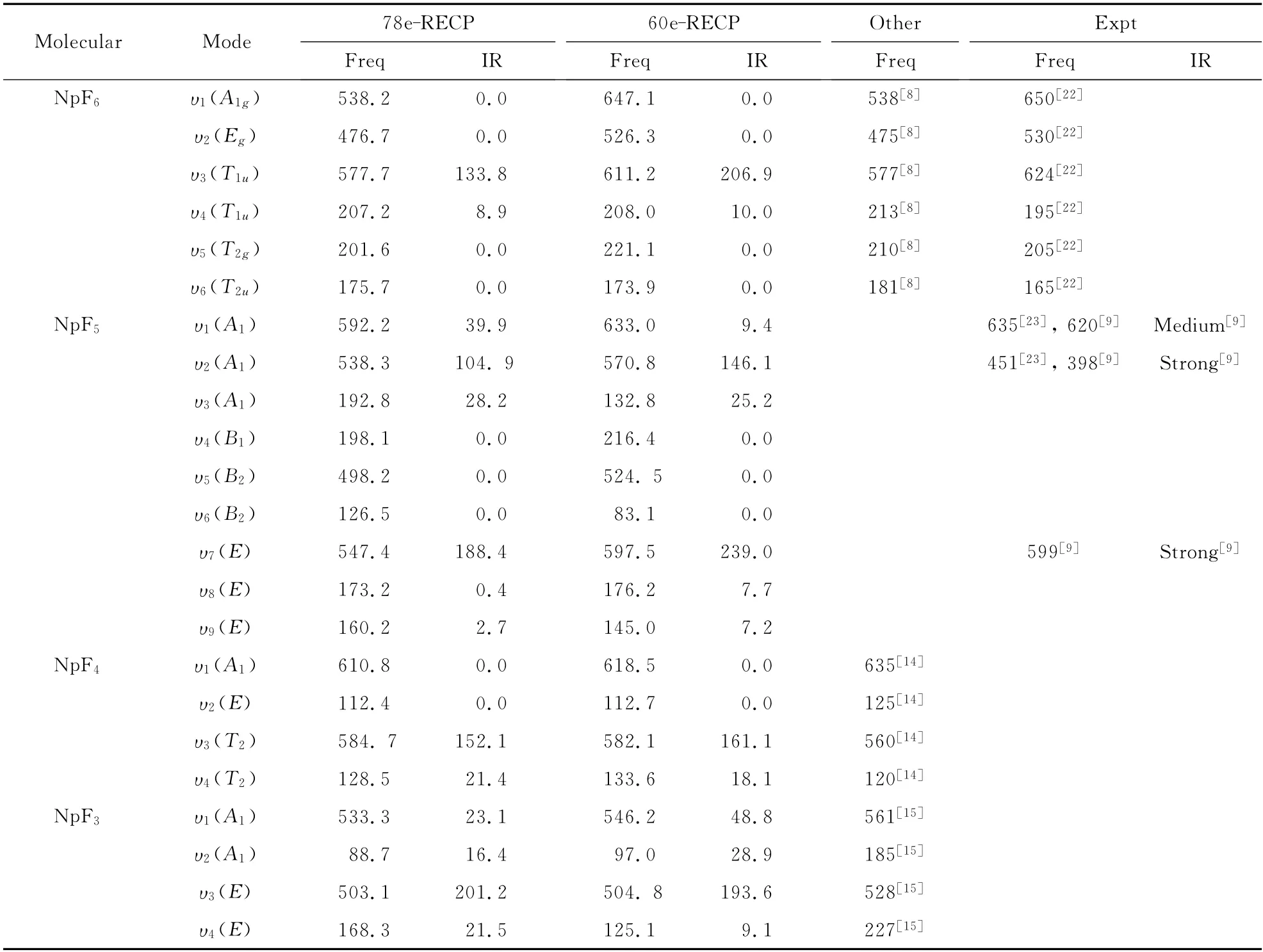
表2 NpFn分子的振动频率/cm-1和IR 强度/(KM/Mole)Table 2 Vibrational harmonic frequencies/cm-1 and infrared intensities/(KM/Mole)
3.2 NpFn分子的电子结构
为了分析不同芯电子RECP 下分子的电子结构,我们对NpFn分子优化后的结构进行了态密度分析.图2 给出了在60个和78 个芯电子RECP下得到的NpF6分子的总态密度和各元素的分态密度.图中能量在-10eV ~-16eV 之间的几个峰主要由Np的5f轨道和F 的2p轨道组成的能级形成,而在-25eV~-28eV 和-31eV~-37 eV 之间的三个峰包含的能级由Np的6p和F 的2s轨道组成,图中能量最低的两个峰来源于Np的6s轨道的贡献.可以看出,与在78 个芯电子RECP下计算的结果相比,60个芯电子RECP 下Np的6s轨道形成的能级以及Np的6p轨道与F的2s轨道形成的能级均向左边移动,而Np的5f轨道和F的2p轨道形成能级构成的峰变胖.由于RECP中芯电子个数减少,原子的有效核电荷数增加,6s、6p价电子被束缚的更紧,它们反过来又起到了屏蔽作用,使得5f电子的束缚减弱,而F原子由于受到Np 原子中电子重新分布的影响,2s、2p电子的束缚变紧,对F 原子的屏蔽效应变强.与78个芯电子RECP 下电子转移情况相比,Np失去的电子由2.152e变为2.732e,F得到的电子由0.359e变为0.455e,两原子的有效电荷均变大,相互吸引增强,分子的Np-F键长变短.
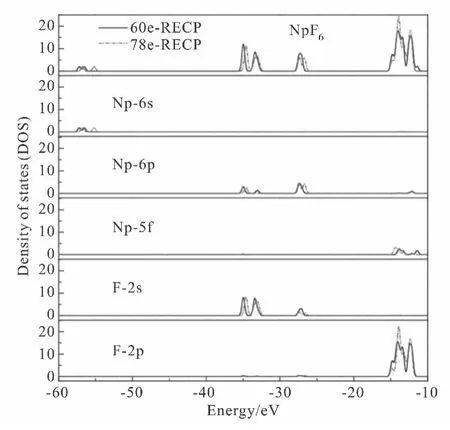
图2 两种芯电子RECP下NpF6分子的总态密度图和分态密度图Fig.2 Total and projected density of states of NpF6 with different RECP
为了进一步讨论NpFn分子的电子结构随F原子个数的变化,图3给出了在60个芯电子RECP下优化后得到的各个分子的总态密度图和分子中电荷的转移情况.图中仅给出了这些分子在-60eV~-4eV 能量范围内各占据轨道的态密度情况.从图可以看出:与NpF6分子相比,分子的总态密度依次整体向右边移动,分子的成键依次减弱.
电负性是用以度量原子对成键电子吸引能力相对大小的量.表3给出了在60个芯电子RECP下优化后得到的各个分子中电荷转移的情况.从表中看出,Np 的电负性小于F,总是失去电子.随着F原子数的减小,Np原子失去的电子依次减少,导致价电子对原子的屏蔽效应逐渐增强,6s、6p和5f电子受到的束缚变小,它们的轨道能升高.单个F原子得到的电子逐渐增多,电子之间的排斥增强,轨道能也升高,比较之下,Np原子的6s、6p和5f电子束缚能抬高的更快,使得对应轨道能差变大,使得Np原子和F 原子的成键依次减弱,结果分子的Np-F键长逐渐增长.

图3 NpFn分子的总态密度随F原子个数的变化Fig.3 Total density of states of NpFn with different n

表3 NpFn分子的电子转移Table 3 The charge transfer in NpFn molecules
4 结 论
本文采用杂化密度泛函(B3LYP)方法,在两种不同芯电子RECP 下,系统优化了NpFn(n=3~6)分子的几何结构,得到了分子的结构参数和振动频率,将计算结果与已有的理论和实验值进行了比较,并对优化的结构进行了NBO 分析.同时,详细讨论了不同芯电子RECP 下分子的电子结构,发现在60个芯电子RECP下,Np原子和F原子受到的屏蔽效应增强,有效电荷增加,从而导致了分子的Np-F键长减小.通过对NpFn系列分子总态密度的比较发现:随着与Np 结合的F原子数目的减少,Np原子对6s、6p和5f电子的束缚减弱,但F 原子对电子的缚减弱的更快,导致成键效果逐步越弱,分子的键长依次增大.
[1] Zhou P D.the prediction of minor actinides amounts accumulated in the spent fuel in china[J].Chinese Journal of Nuclear Science and Engineering,2000,20:11(in Chinese)[周培德.我国核电发展中少数锕系核素积累量预测[J].核科学与工程,2000,20:11]
[2] Zhu Z H,Ming D Q.The delocalization effect of5f electrons for the actinide elements Th to Es[J].Acta Phys.Sin.,2011,60(4):040301(in Chinese)[朱正和,蒙大桥.锕系元素钍到锿的5f电子效应[J].物理学报,2011,60(4):040301]
[3] Kirker I,Kaltsoyannis N.Does covalency really increase across the 5fseries?A comparison of molecular orbital,natural population,spin and electron density analyses of AnCp3(An=Th-Cm;Cp=η5-C5H5)[J].Dalton Trans.,2011,40(1):124.
[4] Pantazis D A,Neese F.All-electron scalar relativistic basis sets for the actinides[J].J.Chem.Theory Comput.,2011,7:677.
[5] de Jong W A,Harrison R J,Nichols J A,et al.Fully relativistic correlated benchmark results for uranyl and a critical look at relativistic effective core potentials for uranium [J].Theor.Chem.Acc.,2001,107:22.
[6] Kaltsoyannis N,Bursten B E.Electronic structure of f1actinide complexes.1.nonrelativistic and relativistic calculations of the optical transition energies of AnXq-6complexes[J].Inorg.Chem.,1995,34(10):2135.
[7] Kimura M,Schomaker V,Smith D W,et al.Electron-Diffraction investigation of the hexafluorides of tungsten,Osmium,Iridium,Uranium,Neptunium,and Plutonium [J].J.Chem.Phys.,1968,48(9):4001.
[8] Hay P J,Martin R L.Theoretical studies of the structures and vibrational frequencies of actinide compounds using relativistic effective core potentials with Hartree-Fock and density functional methods:UF6,NpF6,and PuF6 [J].J.Chem.Phys.,1998,109(10):3875.
[9] Brown D,Whittaker B,Berry J A,et al.The preparation and some properties of actinide pentafluorides[J].J.Less-Common Metals.,1982,86:75.
[10] Eller P G,Asprey L B,Kinkead S A,et al.Reactions of dioxygen difluoride with neptunium oxides and fluorides[J].J.Alloys Comp.,1998,269:63.
[11] Moritz A,Dolg M.Quasirelativistic energy-consistent 5f-in-core pseudopotentials for pentavalent and hexavalent actinide elements [J].Theor.Chem.Account.,2008,121:297.
[12] Moritz A,Cao X Y,Dolg M.Quasirelativistic energy-consistent 5f-in-core pseudopotentials for trivalent actinide elements [J].Theor.Chem.Acc,2007,117:473.
[13] Ellis D E,Rosén A,Gubanov V A.Electronic structure of tetrafluoro-and tetraoxo-actinide complexes[J].J.Chem.Phys.,1982,77(8):4051.
[14] Konings R J M,Hildenbrand D L.The vibrational frequencies,molecular geometry and thermodynamic properties of the actinide tetrahalides[J].J.Alloys.Comp.,1998,271-273:583.
[15] Kahn L R,Baybutt P,Truhlar D G.Ab initio effective core potentials:reduction of all-electron molecular structure calculations to calculations involving only valence electrons[J].J.Chem.Phys.,1976,65(10):3826.
[16] http://www.theochem.uni-stuttgart.de/pseudopotentials
[17] Lee C,Yang W,Parr R G.Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density[J].Phys.Rev.B,1988,37(2):785.
[18] Becke A D.Density-functional thermochemistry.Ⅲ.The role of exact exchange [J].J.Chem.Phys.,1993,98(7):5648.
[19] Frisch M J,Trucks G W,Schlegel H B,et al.Gaussian 03.Revision E.01.Wallingford CT:Gaussian Inc.2004.
[20] Weinstock B,Goodman G L.Vibrational properties of hexafluoride molecules[J].Adv.Chem.Phys.,1965,9:169.
[21] Chayhorsky A A.Chemistry of neptunium [M].Translated by Qiu X X.Beijing:Atomic Energy Press,1978(in Chinese)[柴霍尔斯基A A.镎化学[M].邱孝喜,译.北京:原子能出版社,1978]
[22] Person W B,Kim K C,Campell G M,et al.Absolute intensities of infrared-active fundamentals and combination bands of gaseous PuF6and NpF6[J].J.Chem.Phys.,1986,85(10):5524.
[23] Drobyshevskii Y V,Serik V F,Sokolov V B.Neptunium oxytetrafluoride and neptunium pentafluoride[Reaction of NpO/sub 2/and NpF/sub 4/with KrF/sub 2/][J].Dokl.Akad.Nauk SSSR,1975,225(5):1079.
