HCOOH 在Pd-Fe(111)(nPd∶nFe=1∶1)表面吸附的密度泛函理论研究
2014-07-13张福兰徐伯华徐建华胡武洪
张福兰,徐伯华,徐建华,胡武洪
(长江师范学院化学化工学院,涪陵408100)
1 引 言
随着能源的不断短缺,寻找新型且清洁能源成为当今各科研工作者们的重要课题.在新能源开发领域中,具有能量转化率高、污染小等特点的燃料电池已成为人们的研究热点之一[1].根据使用的电解质,燃料电池可分为碱性燃料电池、磷酸燃料电池、固体氧化物燃料电池、熔融碳酸盐燃料电池、质子交换膜燃料电池,甲酸燃料电池等几大类.其中,直接甲酸燃料电池作为一种高效、清洁、稳定的电源技术,在航天航空以及军事领域都得以成功应用,引起了广泛的关注.目前,主要用于直接甲酸燃料电池的甲酸氧化电催化剂有Pt,Pd,Rh,Ir和Pt基复合催化剂,其中,研究发现Pd对甲酸氧化的电催化活性较高[2].但是,Pd是贵金属,纯Pd催化剂的成本很高,于是人们就往Pd金属中掺杂非贵重金属Fe,Co,Ni,Cr等形成合金催化剂,而且,已有研究发现此合金催化剂的催化效果接近贵金属的催化效果[3-7].其中,冯兰英等[7]以Fe为掺杂元素,采用液相浸渍还原法制备了Pd∶Fe原子比分别为1∶1,2∶1,1∶2 的Pd-Fe/C 催化剂,并进行了相关表征和性能测试,并重点考察了3种不同比例催化剂对甲酸的电催化氧化性能,结果表明Pd/Fe原子比为1∶1时催化性能最好.本研究小组采用密度泛函理论与周期性平板模型相结合的方法,研究过小分子在金属表面的吸附性能[8-10],并 得 到 了 很 好 的 结 果.为 了 弄 清 楚HCOOH 在催化剂表面的作用机制,本文运用密度泛函理论和周期平板模型相结合的方法,模拟研究了HCOOH 在Pd-Fe(111)表面(按原子个数nPd∶nFe=1∶1 的比例在Pd元胞中掺杂Fe原子截面得到)的吸附行为,以此为实验结果提供相应的理论依据.
2 计算模型与方法
采用密度泛函理论和周期性平板模型相结合的方法模拟计算了HCOOH 在Pd(111)和Pd-Fe(111)表面的吸附情况.采用Michaelides等[11]研究CH2和H 在Ni(111)表面吸附类似的方法和模型,取3×3×3三层原子的平板周期性模型模拟Pd(111)表面和Pd-Fe(111)表面,如图1 所示,(a),(b),(c),(d)分别是Pd(111)和Pd-Fe(111)的真空周期表面模型的侧视图和俯视图,真空层厚度为2.0nm.计算过程中考虑表面弛豫,固定下面两层原子,让与吸附物种最近层金属原子自由移动;对C,O 和H 原子采用全电子基组,而对Pd原子和Fe原子采用有效核势(ECP)赝势;函数采用广义梯度近似(GGA)中Perdew-Wang-91(PW91)函数;Quality设为fine,优化收敛精度取程序内定值,在布里渊区积分中k 点设置为4×4×1.HCOOH 在Pd(111)和Pd-Fe(111)表面的吸附位如图2所示.吸附能通过下面方程计算得到:

其中Eads和Es分别代表吸附前HCOOH,Pd(111)和Pd-Fe(111)的能量,Eads/s代表吸附HCOOH 后体系的总能量.全部计算工作使用Materials Studio 4.0版本中的Dmol3模块[12,13].
3 结果与讨论
3.1 平衡构型
表1 列出了HCOOH 在Pd(111)和Pd-Fe(111)表面各吸附位的吸附能.图3是HCOOH 和HCOOH 吸附在Pd(111)、Pd-Fe(111)表面最佳吸附位的稳定构型示意图,其中括号中的数据为各原子的密立根电荷.
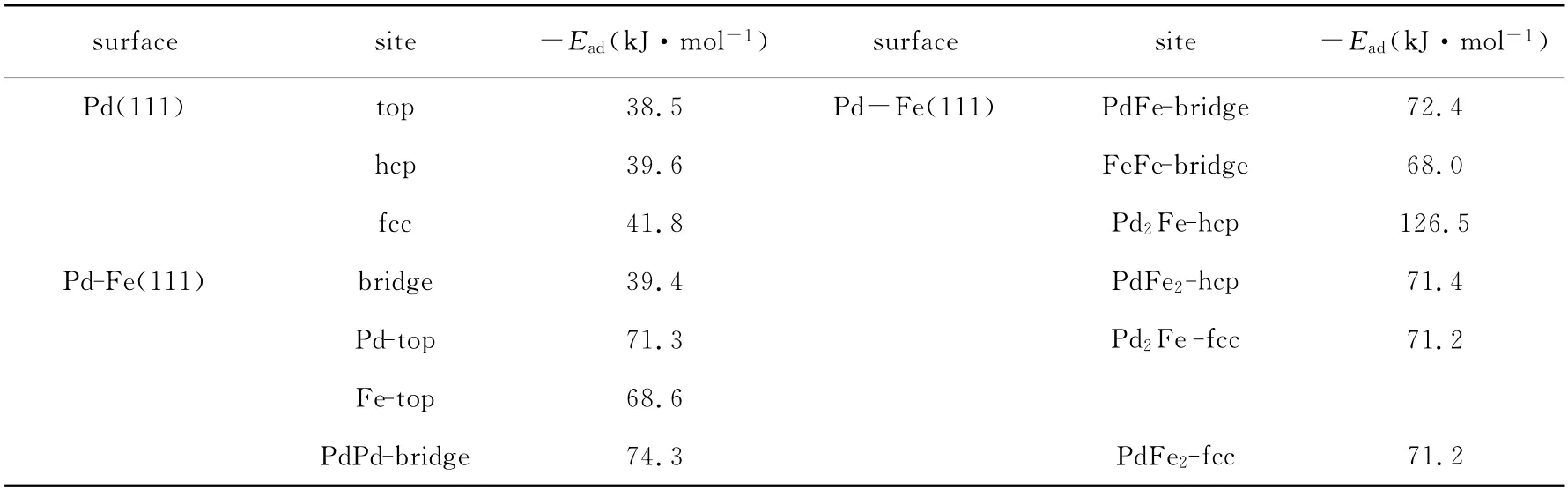
表1 HCOOH 在Pd(111)和Pd-Fe(111)表面各吸附位的吸附能(Ead/kJ·mol-1)Table 1 Adsorption energies(Ead)of the HCOOH on Pd(111)and Pd-Fe(111)surfaces

图1 Pd(111)和Pd-Fe(111)表面周期性平板模型Fig.1 The surfaces periodic slab models of Pd(111)and Pd-Fe(111)
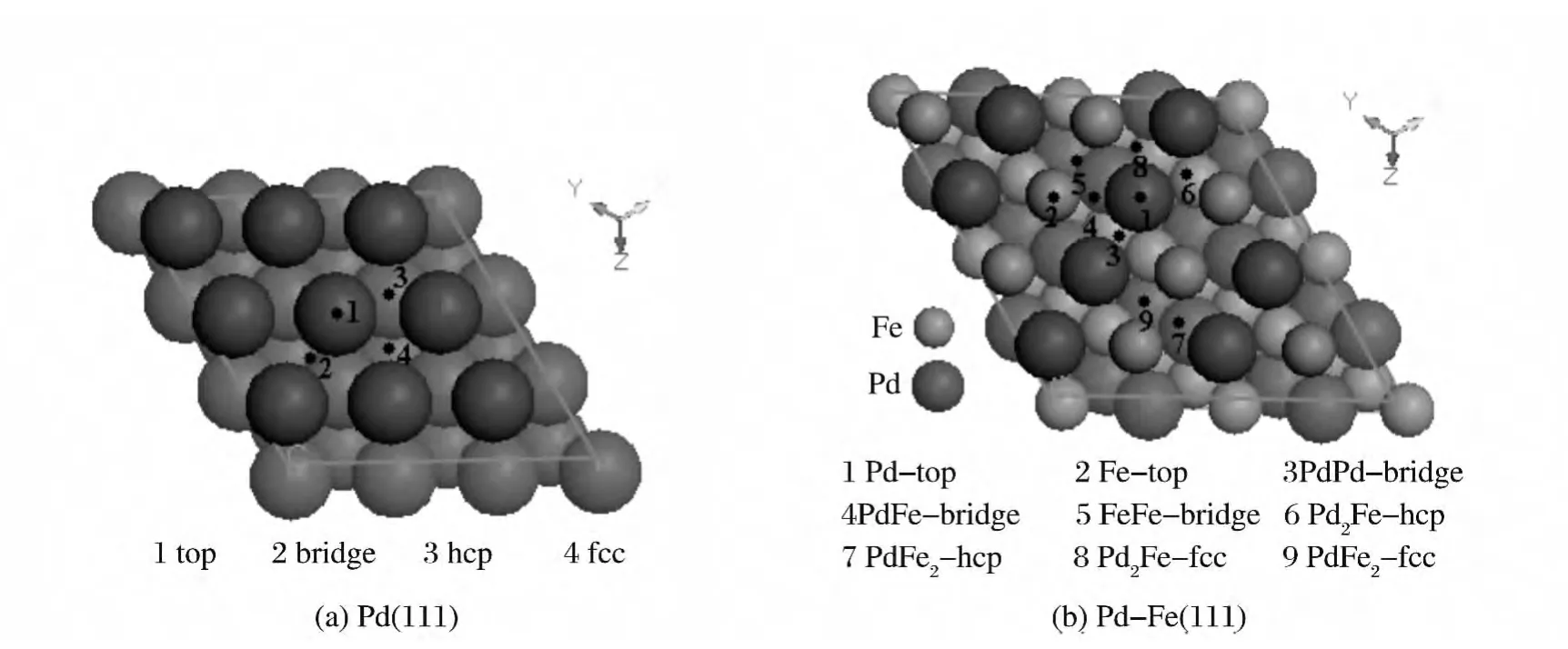
图2 Pd(111)和Pd-Fe(111)表面的吸附位Fig.2 The model of adsorption sites on Pd(111)and Pd-Fe(111)surfaces
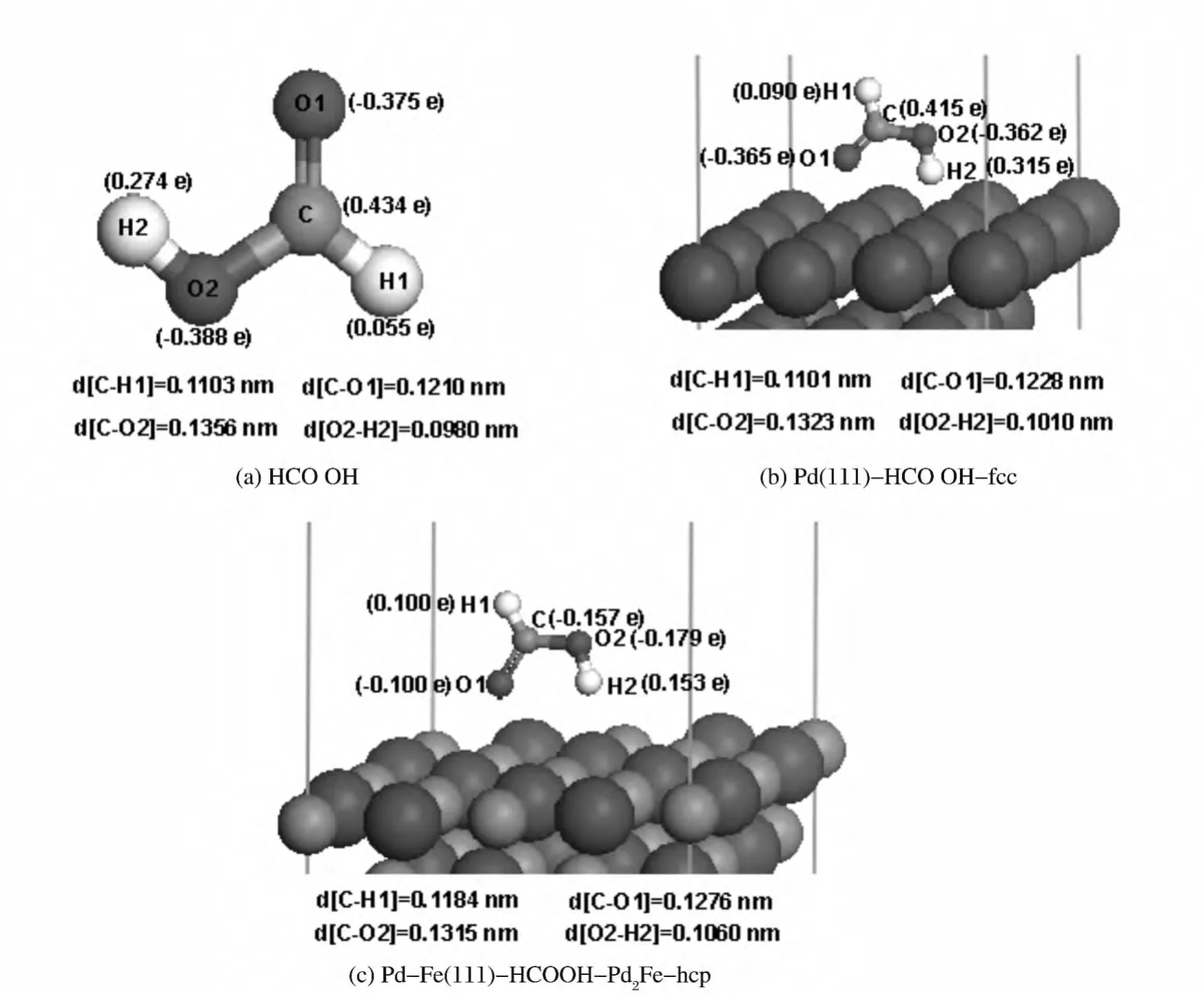
图3 HCOOH 在Pd(111)和Pd-Fe(111)表面最佳吸附位的构型示意图Fig.3 The equilibrium adsorption of theHCOOH on the most stable site of Pd(111)and Pd-Fe(111)surfaces
由表1 知,HCOOH 在Pd(111)表面top,bridge,hcp,fcc四个位的吸附能分别为-38.5,-39.4,-39.6,-41.8kJ·mol-1,其中fcc位的吸附能的绝对值最大,为最佳吸附位,构型参数见图3(b).计算得到气相HCOOH 的分子结构的构型参数见图3(a).由图3(a)可知,计算得到气相HCOOH 分子中C-H1键长为0.1103nm,O2-H2键长为0.0980nm,C=O1键长为0.1210nm,C-O2键长为0.1356nm.当HCOOH 吸附在Pd(111)表面fcc位时,C-H1,O2-H2,C=O1,C-O2的键长分别为0.1101nm,0.1010nm,0.1228nm,0.1323nm,数据显示吸附过程中CH1,O2-H2,C=O1三键分别拉长,而C-O2缩短,而且两个C-O 键长也趋于平均化,说明吸附过程中,两个H 原子趋向于脱离HCOOH 分子而生成产物CO2,此结论和冯兰英等[7]的实验研究结果一致.密立根电荷分析表明,在气相HCOOH分子中,C,H1,H2,O1,O2所带密立根电荷分别为0.434 e,0.055 e,0.274 e,-0.375 e,-0.388 e, 整 个 HCOOH 分 子 显 中 性.当HCOOH 吸附到Pd(111)表面后,C,H1,H2,O1,O2所带密立根电荷分别为0.415e,0.090e,0.315e,-0.365e,-0.362e,说明吸附过程中,两个H 原子的正电性增加,有利于脱离分子;而且数据显示整个HCOOH 分子带0.093e的正电荷,表明吸附过程中HCOOH 向Pd(111)表面转移了电子.由此电荷和结构的改变说明HCOOH 在Pd(111)表面为化学吸附.
又由表1知,HCOOH 在Pd-Fe(111)表面Pd-top,Fe-top,PdPd-bridge,PdFe-bridge,FeFebridge,Pd2Fe-hcp,PdFe2-hcp,Pd2Fe-fcc,PdFe2-fcc 等9 个吸附位的吸附能分别为-71.3,-68.6,-74.3,-72.4,-68.0,-126.5,-71.4,-71.2,71.2kJ·mol-1,其中Pd2Fe-hcp位的吸附能的绝对值最大,为最佳吸附位,构型参数见图3(c).当HCOOH 吸附在Pd-Fe(111)表面Pd2Fe-hcp位时,C-H1,O2-H2,C=O1,CO2的键长分别为0.1184nm,0.1276nm,0.1060 nm,0.1315 nm,数据显示,与图3(b)对比,HCOOH 吸 附 在Pd-Fe(111)表 面Pd2Fe-hcp 位时,键长变化得更多,中C-H1,O2-H2两键拉得更长,C=O1和C-O2两键更平均化,说明形成CO2的趋势更强了,这和HCOOH 分子吸附在Pd-Fe(111)表面的吸附能大于吸附在Pd(111)表面的吸附能结论一致,且此结论和冯兰英等[7]的实验研究结果一致.密立根电荷分析表明,当HCOOH 吸附到Pd-Fe(111)表面后,C,H1,H2,O1,O2所带密立根电荷分别为-0.157e,0.100 e,0.153e,-0.179e,-0.100e,说明吸附过程中,两个H 原子的正电性增加,有利于脱离分子;而且数据显示整个HCOOH 分子带-0.183e的电荷,表明吸附过程中Pd-Fe(111)表面向HCOOH 转移了电子.由此电荷和结构的改变说明HCOOH 在Pd-Fe(111)表面为化学吸附.
3.2 电子结构
图4给出了清洁表面Pd(111)和Pd-Fe(111)的能带和态密度(DOS)图.由图4 可知,Pd(111)表面的能带区域为-9.54 ~-0.01eV,Pd-Fe(111)表面的能带区域为-8.70~-0.68eV,Pd金属掺杂Fe后,催化剂的能带变窄了1.53eV,但是峰高增加,说明电子云密度增加,有利于电荷转移,说明催化剂活性增加,这和实验研究结果一致.

图4 清洁表面Pd(111)和Pd-Fe(111)的能带和态密度(DOS)图(EF为费米能级,0.0eV)Fig.4 Energy band structures and densities of states(DOS)for the clean Ru(001)and Ru-Co(001)surfaces.The Fermi levels(EF)of each system are aligned to 0.0eV
4 结 论
采用周期性平板模型与密度泛函理论相结合的方法,对HCOOH 在Pd(111)表面top,fcc,hcp,bridge 四个吸附位和Pd-Fe(111)表面Pdtop,Fe-top,PdPd-bridge,PdFe-bridge,FeFebridge,Pd2Fe-hcp,PdFe2-hcp,Pd2Fe-fcc,Pd-Fe2-fcc等9个吸附位的13 种吸附模型进行了能量计算、构型优化,得到了HCOOH 较有利的吸附位.结果表明:HCOOH 在Pd(111)表面的最稳定吸附位为fcc位,其吸附能的大小顺序为fcc﹥hcp﹥bridge﹥top;HCOOH 在Pd-Fe(111)表面的最稳定吸附位为Pd2Fe-hcp位,其吸附能的大小顺序为Pd2Fe-hcp﹥PdPd-bridge﹥PdFe-bridge﹥PdFe2-hcp﹥Pd-top﹥Pd2Fe-fcc=PdFe2-fcc﹥FeFe-bridge﹥Fe-top;Fe 作为掺杂元素,Pd-Fe(111)催化剂有更好的催化活性,HCOOH 在金属表面属于化学吸附.
[1] Chen Z X,Huang Y C,Li Z,et al.Theoretical Chemistry and New Energy Sources[J].Progress in Chem.,2009,21(11):2271(in Chinese)[陈兆旭,黄玉成,李哲,等.理论化学与新能源[J].化学进展,2009,21(11):2271]
[2] Yuan Q Y,Tang Y W,Zhou Y M,et al.Formic acid as methanol-alternative fuel in direct methanol fuel cell chinese[J].J.Appl.Chem.,2005,22(9):929(in Chinese)[袁青云,唐亚文,周益明,等.甲酸作为直接甲醇燃料电池替代燃料[J].应用化学,2005,22(9):929].
[3] Tamizhmani G,Gapuano G A.Improved electrocatalytic oxygen reduction performance of platinum ternary alloy-oxide in SPFCs[J].J.Electrochem Soc.,1994,14(4):968.
[4] Lee K,Savadogo O,Ishihara A,et al.Methano1-tolerant oxygen reduction electrocatalysts based on Pd-3dtransition metalalloys for direct methanol fuel cells[J].J.Electrochem.Soc.,2006,153(1):20.
[5] Mustain W E,Kepler K,Prakash J.Investigations of carbon supported CoPd3catalysts as oxygen cathodes in PEMFCs[J].Electrochemistry Communications,2006(8):406.
[6] Shao M H,Sasaki K,Adzic R R.Pd-Fe Nanoparticles as electrocatalysts for oxygen reduction[J].J.Amer.Chem.Soc.,2006,128:3526.
[7] Feng L Y,Jiang H,Zhu H,et al.Effect of Fe content on the performance of Pd/C catalyst[J],New Chem.Mat.,2008,36(1):45(in Chinese)[冯兰英,江红,朱红,等.Fe含量对Pd/C 催化剂性能的影响[J].化工新型材料,2008,36(1):45]
[8] Zhang F L.Density functional theory study of N2adsorption on Ru(001)surface by doping with cobalt[J].J.At.Mol.Phys.,2010,27(4):769(in Chinese)[张福兰.N2在Co掺杂Ru(001)表面吸附的DFT 研究[J].原子与分子物理学报,2010,27(4):769]
[9] Zhang F L.Density functional theory study of CHx(x=2~4)adsorption on Fe(110)surface[J].J.At.Mol.Phys.,2010,27(5):986(in Chinese)[张福兰.CHx(x=2~4)在Fe(110)表面吸附的DFT研究[J].原子与分子物理学报,2010,27(5):986]
[10] Zhang F L.Density functional theory study of C2Hx(x=4~6)adsorption on Ni(111)surface[J].J.At.Mol.Phys.,2010,27(6):1175(in Chinese)[张福兰.C2Hx(x=4~6)在Ni(111)表面吸附的DFT 研究[J].原子与分子物理学报,2010,27(6):1175]
[11] Michaelides A,Hu P.A density functional theory study of CH2and H adsorption on Ni(111)[J].J.Chem.Phys.,2000,112(13):6006.
[12] Delley B.An all-electron numerical method for solving the local density functional for polyatomic molecules[J].J.Chem.Phys.,1990,92(1):508.
[13] Delley B.From molecules to solids with the DMol3approach.[J].J.Chem.Phys.,2000,113(18):7756.
