抑瘤素M在大鼠非酒精性脂肪性肝病进展中的作用
2014-06-27彭菊聪苌新明张玲娟刘毓屾
彭菊聪,苌新明,张玲娟,刘毓屾,秦 洁
(西安交通大学医学院第一附属医院消化内科,陕西西安 710061)
非酒精性脂肪性肝病(non-alcoholic fatty liver disease, NAFLD)是一种与胰岛素抵抗(insulin resistance, IR)和遗传易感性密切相关,以弥漫性肝细胞大泡性脂肪变性为主要特征的临床病理综合征[1]。随着社会经济的发展,人们生活水平的提高及生活方式的改变,NAFLD的发病率不断攀升,已成为西方发达国家第一大慢性肝病及健康人血清肝酶升高的首要原因[2],在我国也仅次于慢性病毒性肝炎而位居第二,在某些地区甚至超过了病毒性肝炎的发病率。NAFLD发病机制复杂,虽然国内外学者进行了广泛深入的研究,但至今仍然未完全阐明。研究发现,抑瘤素M(oncostatin M, OSM)可促进IL-6和TNF-α表达[3-4],能抑制脂联素(adiponectin, ADP)的产生[5],还能介导IR形成[6]。IR、IL-6、TNF-α、ADP又与NAFLD发病密切相关,但有关OSM及受体(OSM receptor, OSMR)与NAFLD关系的研究报道甚少。因此,研究OSM与OSMR在NAFLD中的表达变化,有助于进一步了解NAFLD可能的发病机制,并为诊断治疗提供新的实验依据。
1 材料与方法
1.1实验动物健康雄性SD大鼠30只,清洁级,体质量(140±20)g,购自西安交通大学医学院实验动物中心。
1.2主要原料及试剂胆固醇(中国医药集团上海化学试剂公司),胆酸钠(北京奥博星生物有限责任公司)。大鼠OSM ELISA试剂盒(96人份,美国RD Biosciences公司)。兔抗大鼠OSM和OSMR多克隆抗体(北京博奥森生物技术有限公司),即用型快捷免疫组化MaxVisionTM试剂盒(福州迈新生物技术开发有限公司)。
1.3NAFLD模型的建立及取材30只雄性SD大鼠适应性喂养1周后,用随机数字表法随机分为正常对照组和模型组,各组15只。对照组给予普通饲料,模型组饲予高脂饮食(配方组份比例为:82.5%标准饲料+2%胆固醇+10%猪油+5%蛋黄粉+0.5%胆酸钠[7])。所有实验大鼠均自由饮水进食和活动,分笼饲养在本校动物中心普通动物房,共饲养12周。
12周末隔夜禁食后处死所有大鼠,处死前称体质量。以100 g/L水合氯醛(0.3~0.5 mL/100 g)麻醉,腹主动脉取血5~6 mL,以备血清OSM、胰岛素及其他生化指标的检测。取血后迅速摘取完整肝脏,称肝湿重并计算肝指数:肝指数=肝湿重(g)/体质量(g)×100%。取每只大鼠肝右叶相同部位1.0 cm×1.0 cm×0.5 cm大小的两块肝组织,一部分立即行冰冻切片,另一部分用40 g/L多聚甲醛固定。
1.4血清生化指标的测定采用自动生化分析仪检测大鼠血清丙氨酸氨基转移酶(ALT)、天门冬氨酸氨基转移酶(AST)、总胆固醇(CHOL)、甘油三酯(TG)和空腹血糖(fasting plasma glucose, FPG)。用放射免疫法检测空腹胰岛素(fasting serum insulin, FINS)水平,用HOMA-IR方法计算胰岛素抵抗指数(IRI),IRI=(FPG×FINS)/22.5。ELISA双抗夹心法检测血清OSM含量。
1.5肝组织病理学检查肝组织冰冻切片行油红O染色,石蜡切片行HE染色,光镜下观察肝脏组织学变化。NAFLD活动度积分标准参照2010年版《非酒精性脂肪性肝病诊疗指南》[1]。
1.6免疫组织化学检测采用Max VisionTM二步法染色肝组织OSM和OSMR,具体操作步骤按MaxVisionTM试剂盒说明书进行。5 μm厚组织切片经柠檬酸缓冲液微波修复抗原;30 mL/L H2O2孵育灭活内源性过氧化物酶;兔抗大鼠多克隆抗体(OSM按1∶100稀释,OSMR按1∶150稀释)孵育后滴加MaxVisionTM试剂,显色、封片。
免疫组化结果判定:由2位病理医师采用双盲法评估结果。每张切片均随机选取5个互不重叠的高倍视野(×400)。综合阳性细胞比例和染色强度进行半定量分析。阳性细胞所占百分率计分:无阳性细胞0分;阳性细胞<10%,1分;10%~50%,2分;>50%,3分。染色强弱计分:不着色,0分;淡黄色,1分;黄色,2分;棕黄色,3分。两项得分相加为免疫组化评分。

2 结 果
2.1大鼠体质量、肝湿重及肝指数的变化大鼠体质量随着造模吋间延长不断增加,12周末模型组大鼠体质量高于对照组,但差异无统计学意义(P=0.438)。模型组肝湿重、肝指数显著高于对照组(P<0.01,表1)。
表1两组大鼠体质量、肝湿重及肝指数的比较
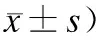

与对照组比较,**P<0.01。
2.2大鼠肝功、血脂的变化模型组血清ALT、AST和CHOL较正常对照组显著升高,差异有统计学意义(P<0.01),模型组血清TG较正常对照组升高,差异无统计学意义(P>0.05,表2)。
2.3大鼠血清OSM、FINS和IRI的变化与对照组相比,模型组血清OSM、FINS明显升高,差异有统计学意义(P<0.05),FPG及IRI显著升高(P<0.01,表2)。
表2两组大鼠血清生化指标的比较

与对照组比较,*P<0.05;**P<0.01。
2.4肝脏大体及病理学改变对照组大鼠肝脏表面光滑,边缘锐利,颜色红润有光泽。HE染色见肝小叶结构完整、清晰,肝细胞基本无脂肪变性,油红O染色见少量体积较小的红色脂滴。模型组大鼠肝脏体积显著增大,边缘钝圆,颜色呈土黄色,切面油腻感。HE染色见肝小叶结构欠清晰,肝窦间隙明显变窄,肝细胞胞质内出现大小不等的圆形空泡,严重者细胞核被脂滴挤向一侧,小叶内及汇管区可见局灶性坏死、碎屑样坏死和点片状炎性细胞浸润。油红O染色见肝细胞胞质内充满了鲜红色脂滴。模型组肝组织脂肪变性、炎症及气球样变程度较对照组严重,差异有统计学意义(P<0.01)。模型组肝组织NAFLD活动度积分都在5分以上,平均积分大于4分,因此模型组大鼠均达NASH诊断标准;对照组大鼠NAFLD活动度积分均小于3分,为正常肝组织(图1、表3)。

图1两组大鼠肝脏病理学变化
Fig.1 Hepatic histopathological changes between two group rats
A、C:对照组;B、D:模型组。A、B:HE,×400;C、D:油红O染色,×200。
表3大鼠肝组织NAFLD活动度积分的比较
Tab.3 Comparison of NAFLD activity scores between the two groups (n=15)
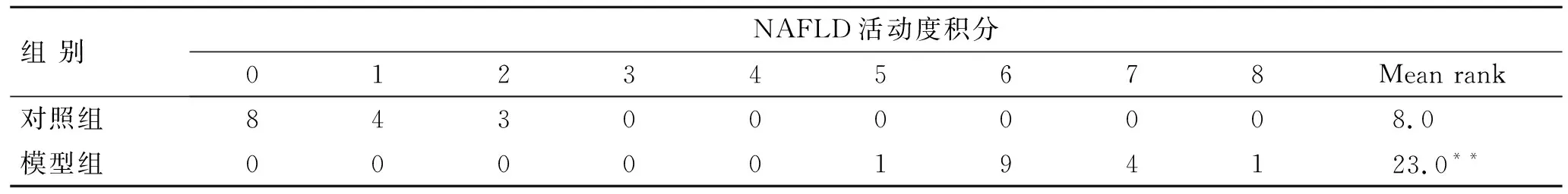
与对照组比较,**P<0.01。
2.5肝脏OSM和OSMR的表达结果OSM主要表达于浸润的炎症细胞和胆管上皮细胞胞膜及胞质,枯否细胞(Kupffer cells, KC)及肝细胞胞膜及胞质也见少量表达。模型组OSM表达评分(3.93±1.09)显著高于对照组(2.80±0.77)(P<0.01,图2)。OSMR主要表达在肝细胞胞质及胞膜,尤其是中央静脉周围的肝细胞。对照组OSMR表达评分(2.33±1.04)低于模型组(4.0±1.31),差异有统计学意义(P<0.01,图3)。
3 讨 论
NAFLD是一种慢性低度炎症和IR状态,IR是NAFLD的重要特征。1998年DAY等[8]所提出了NAFLD发病机制的“二次打击”学说,认为IR是NAFLD发病的中心环节,已被广泛认可。研究还表明,IR还是NAFLD发病的始动因素而非结果,贯穿于发病机制全过程[9]。SAKURAI等[10]研究发现,IR与肝脏脂肪变性的相关性较体脂指数高,几乎所有的NAFLD患者存在肝脏和周围组织的IR,而不一定存在肥胖及糖脂代谢紊乱。这都说明IR是NAFLD的一个极其重要的独立危险因素。本实验模型组FPG较对照组显著升高,FINS和IRI也较对照组明显升高,差异有统计学意义。说明模型组大鼠胰岛素敏感性下降,存在IR、高胰岛素血症状态,这与既往研究结果一致。

图2OSM在各组肝脏组织中的表达
Fig.2 The positive expression of OSM in the liver tissue of each group (×400)
A、B:对照组;C、D:模型组。

图3OSMR在各组肝脏中的表达
Fig.3 The positive expression of OSMR in the liver tissue of each group (×400)
A:模型组;B:对照组。
OSM是一种能耐酸、耐热的分泌型单链糖蛋白。最初是ZARLING等[11]于1986年从佛波醇12-肉豆蔻酸盐13-乙酸酯((phorbol-12-myristate-13-acetate, PMA)培育的淋巴瘤细胞U937细胞上清分离提纯获得,因具有抑制A351黑色素瘤细胞系生长作用而得名。随着研究的深入,发现OSM还能调节炎症反应,参与肥胖、动脉粥样硬化、2型糖尿病等代谢性疾病的发生发展[3-4,12-13]。OSM由单核细胞-巨噬细胞、中性粒细胞、嗜酸性粒细胞、活化的T细胞等分泌[14-15]。肝脏中的KC也可分泌OSM[6]。OSM有两种类型受体,Ⅰ型OSMR和Ⅱ型OSMR,Ⅱ型为OSM的特异性受体。鼠OSM也有两种受体,但只与Ⅱ型OSMR结合,不与Ⅰ型OSMR结合。
本研究结果显示,OSM在对照组大鼠血清和肝组织低表达,而在模型组表达显著增高,这与HENKEL等[6]在小鼠NAFLD模型中的研究结果一致。HENKEL还发现OSM的升高早于IL-6,升高水平也较IL-6显著,在肝脏未出现明显炎症前即有明显升高。因此推测OSM升高是NAFLD发病的早期事件而非结果。此外,OSM可诱导星形细胞和成纤维细胞表达IL-6,OSM促进IL-6的分泌呈时间和剂量依赖性;BAKER等[3]研究发现,OSM可通过激活NF-κB信号途径诱导TNF-α的表达。IL-6和TNF-α可促进脂肪分解、炎症反应,诱导细胞凋亡,介导IR形成,为NAFLD重要致病因子,与NAFLD的严重程度正相关。SONG等[5]研究发现,OSM可抑制脂肪细胞ADP mRNA表达和蛋白的分泌,ADP可增强胰岛素的敏感性、减轻肝脏脂肪沉积和抗炎抗肝纤维化作用,是NAFLD的重要保护因素。还有研究发现,OSM可增加肝细胞细胞因子信号转导抑制因子3(suppressor of cytokine signaling3, SOCS3)mRNA表达,增高的SOCS3又可阻碍胰岛素受体底物IRS-1和IRS-1的酪氨酸磷酸化,干扰胰岛素信号通路,介导IR[6,16]。此外,OSM也可直接抑制胰岛素依赖的Akt激酶的磷酸化和调节葡萄糖降解和糖原合成的葡萄糖激酶的活性,使葡萄糖利用发生障碍,导致IR。这提示OSM促进了NAFLD的进展。OSM主要表达于浸润的炎症细胞、胆管上皮细胞胞膜及胞质,KC和肝细胞胞膜及胞质也见少量表达。这与既往研究所发现的OSM可由多种细胞分泌,OSM广泛表达于炎症反应和损伤组织修复部位结果基本一致[14-15,17]。目前尚未发现有关OSMR与NAFLD发病关系的研究报道。由于大鼠虽有两种OSMR,但研究发现只与特异性的Ⅱ型受体结合,因此本实验只检测了OSM的特异性受体。OSMR在模型组肝组织表达较对照组明显升高,差异有统计学意义。OSMR主要在肝细胞胞质及胞膜表达,尤其是中央静脉周围的肝细胞。这与NOYKO等[18]在肝硬化患者中检测到OSMR的表达部位一致。NAKAMURA等[19]研究发现,OSMR在正常小鼠肝组织基本不表达,而给以CCL4诱导肝脏损伤后OSMR表达增高,认为OSMR参与了组织损伤反应。
综上所述,循环血中OSM和肝组织中OSM、OSMR表达升高参与了NAFLD发病,OSM可能是NAFLD发病的一个危险因子。但是OSM及OSMR表达升高的原因及在NAFLD发病网络机制中的确切作用有待进一步研究。
参考文献:
[1] 中华医学会肝脏病学分会脂肪肝和酒精性肝病学组. 非酒精性脂肪性肝病诊疗指南[J]. 中国肝脏病杂志:电子版, 2010, (4):43-48.
[2] KICHIAN K, MCLEAN R, GRAMLICH LM, et al. Nonalcoholic fatty liver disease in patients investigated for elevated liver enzymes[J]. Can J Gastroenterol, 2003, 17(1):38-42.
[3] BAKER BJ, PARK KW, QIN H, et al. IL-27 inhibits OSM-mediated TNF-alpha and iNOS gene expression in microglia[J]. Glia, 2010, 58(9):1082-1093.
[4] SMYTH DC, KERR C, RICHARDS CD. Oncostatin M-induced IL-6 expression in murine fibroblasts requires the activation of protein kinase Cdelta[J]. J Immunol, 2006, 177(12):8740-8747.
[5] SONG HY, KIM MR, LEE MJ, et al. Oncostatin M decreases adiponectin expression and induces dedifferentiation of adipocytes by JAK3- and MEK-dependent pathways[J]. Int J Biochem Cell Biol, 2007, 39(2):439-449.
[6] HENKEL J, GARTNER D, DORN C, et al. Oncostatin M produced in Kupffer cells in response to PGE2: possible contributor to hepatic insulin resistance and steatosis[J]. Lab Invest, 2011, 91(7):1107-1117.
[7] 钟岚,范建高. 非酒精性脂肪肝动物模型[J]. 国外医学:消化系疾病分册, 1999, 19(3):175-178.
[8] DAY CP, JAMES OF. Steatohepatitis: a tale of twohits?[J]. Gastroenterology, 1998, 114(4):842-845.
[9] KIM HJ, LEE KE, KIM DJ, et al. Metabolic significance of nonalcoholic fatty liver disease in nonobese, nondiabetic adults[J]. Arch Intern Med, 2004, 164(19):2169-2175.
[10] SAKURAI M, TAKAMURA T, OTA T, et al. Liver steatosis, but not fibrosis, is associated with insulin resistance in nonalcoholic fatty liver disease[J]. J Gastroenterol, 2007, 42(4):312-317.
[11] ZARLING JM, SHOYAB M, MARQUARDT H, et al. Oncostatin M: a growth regulator produced by differentiated histiocytic lymphoma cells[J]. Proc Natl Acad Sci U S A, 1986, 83(24):9739-9743.
[12] 李连喜,陶征,梁文昌,等. 抑瘤素M在高血糖致人主动脉平滑肌细胞增殖中的作用[J]. 中华糖尿病杂志, 2009, 1(3):201-205.
[13] LBASANZ-PUIG A, MURRAY J, PREUSCH M, et al. Oncostatin M is expressed in atherosclerotic lesions: a role for Oncostatin M in the pathogenesis of atherosclerosis[J]. Atherosclerosis, 2011, 216(2):292-298.
[14] ORIKAWA Y. Oncostatin M in the development of the nervous system[J]. Anat Sci Int, 2005, 80(1):53-59.
[15] EPOVIC P, BENVENISTE EN. Prostaglandin E2 is a novel inducer of oncostatin-M expression in macrophages and microglia[J]. J Neurosci, 2002, 22(13):5334-5343.
[16] EKI K, KONDO T, KAHN CR. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms[J]. Mol Cell Biol, 2004, 24(12):5434-5446.
[17] UYCKX VA, CAIRO LV, COMPSTON CA, et al. Oncostatin M pathway plays a major role in the renal acute phase response[J]. Am J Physiol Renal Physiol, 2009, 296(4):F875-883.
[18] NOYKO I, SOHARA N, SPICER SS, et al. Expression of oncostatin M and its receptors in normal and cirrhotic human liver[J]. J Hepatol, 2005, 43(5):893-900.
[19] AKAMURA K, NONAKA H, SAITO H, et al. Hepatocyte proliferation and tissue remodeling is impaired after liver injury in oncostatin M receptor knockout mice[J]. Hepatology, 2004, 39(3):635-644.
